There are a great many anabolic steroids which differ from one to another in various (subtle) ways. These differences, in essence, boil down to the underlying differences in chemical structure. In this article I’ll cover some chemical alterations that can be found in common anabolic steroids. I’ll also briefly cover what esters are. Whilst they aren’t a chemical alteration to the parent molecule per se, they do affect an anabolic steroid’s ‘behavior’ regardless because of its effect on the rate of release from the oil into the body.
What is an ester?
The word ester gets thrown around a lot when talking about anabolic steroids. And most people know that it’s something that affects the half-life of the anabolic steroid. But what is it actually?
If you have a molecule, like testosterone, you can attach something to this parent molecule to affect its biophysical properties. In the case of esterification of an anabolic steroid, it boils down to attachment of a carboxylic acid group onto carbon 17 of the steroid backbone as depicted below:
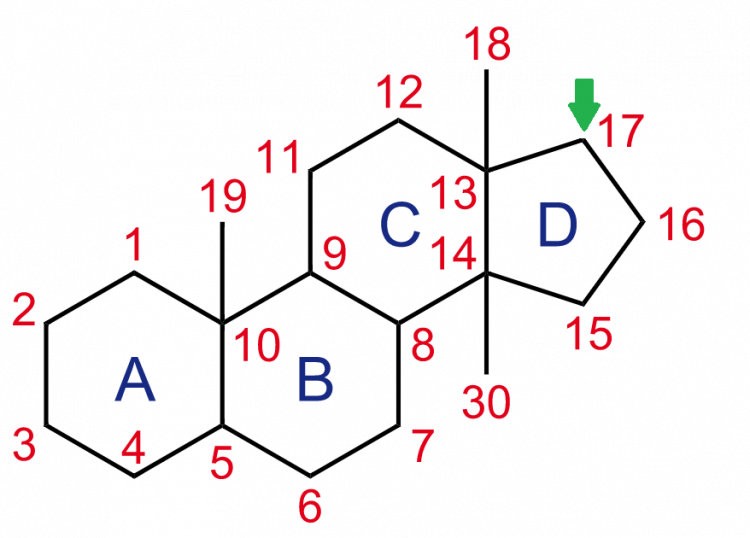
The attachment of this group affects the polarity of the molecule. And the polarity of a molecule relates to how a charge is distributed across it. A charge on a molecule leads to interaction with surrounding charged molecules. This is of particular relevance with regard to water. Water is quite polar and thus molecules which are also quite polar dissolve easily in it. Polar molecules are happy in water.
Lipids, in contrast, are nonpolar, or only slightly so. As a result, they have a hard time dissolving in water. For example, if you drop some olive oil in a glass of water, you can see how the oil sticks together and forms a layer on the water. It isn’t dissolving. This is the result of the lack of polarity of olive oil. Oils aren’t happy in water.
And here comes the crux of the matter: stuff that is nonpolar (or only slightly so) dissolves readily in oil. When an ester is attached to a steroid molecule, it will decrease its polarity and thereby make it more readily dissolvable in oil, and less readily dissolvable in water. Or in other words: it becomes less hydrophilic (‘water-loving’) and more lipophilic (‘oil-loving’). Esterified steroids are happy in oil.
So why is this important? As you might know, esterified steroids have a longer half-life than their nonesterified counterparts. It has the potential to greatly extend the half-life. For example, testosterone undecanoate demonstrated a half-life of 70 days(!) in a small group of hypogonadal men [1]. This long half-life can be directly traced back to the large effect the undecanoate has on the parent molecule’s polarity. It greatly decreases it. So what happens is the following. The esterified testosterone (or any other type of steroid) is dissolved in oil. This oil is then injected intramuscularly in which it will spread across the length of the muscle bundles. This looks as follows (note how the oil spreads along the length of the muscle bundles):
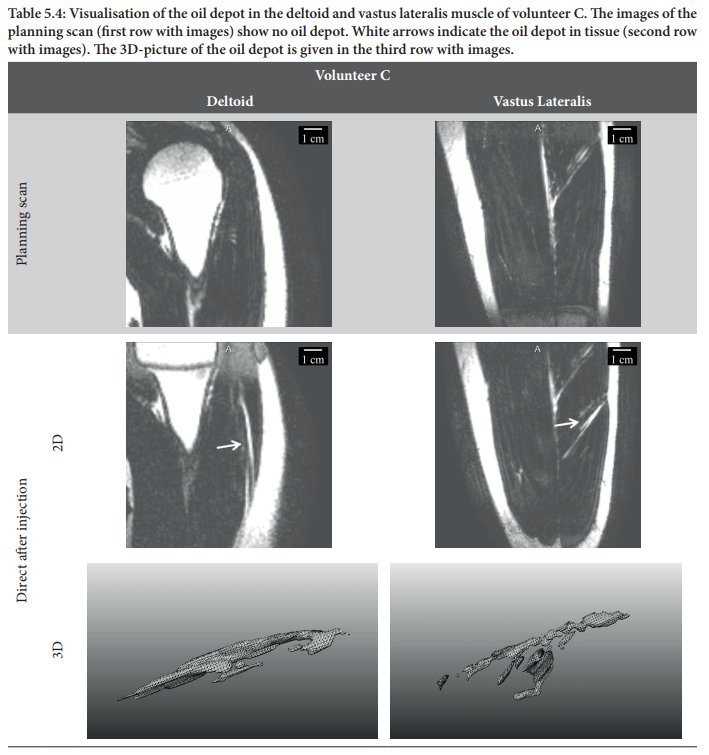
The esterified testosterone will then, to some extent, diffuse out of the oil depot and dissolve in the surrounding tissue, which is made up mostly of water. The rate at which this happens is determined by its polarity. If it’s very nonpolar (and thus lipophilic) it will diffuse very very slowly out of the oil into the surrounding tissue. After all: nonpolar substances are happy in oil, and not happy in water. Thus the esterification dictates the half-life of the injected molecule.
A great rule of the thumb is that, the longer the carbon chain length of the ester, the longer the half-life of the compound will be. Because the longer the chain length, the more it will decrease the polarity of the molecule for reasons I won’t bore you with. In addition to the length of the carboxylic acid, you can also change the structure of it to affect the polarity. For example, this is evident with the ester you see attached to Parabolan: trenbolone hexahydrobenzylcarbonate. Herein you can find a cyclohexane group. I’ve colorized this attachment and highlighted the cyclohexane group in blue:
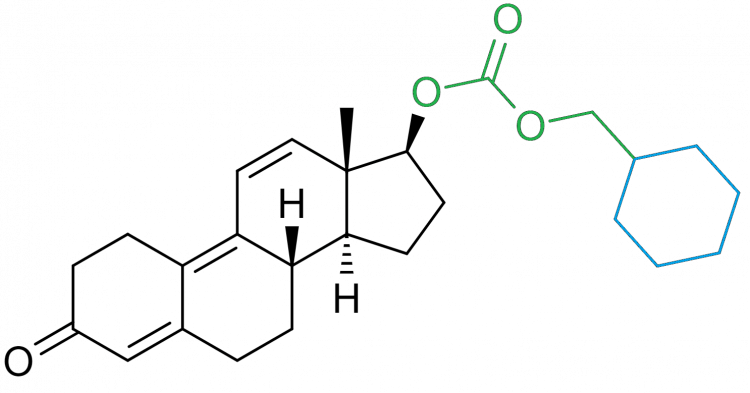
Either way, this is also the reason why testosterone enanthate (6 carbon atoms) has a significantly shorter half-life than testosterone undecanoate (11 carbon atoms) that I’ve mentioned earlier.
Once the esterified steroid hits the bloodstream, the ester is rapidly hydrolyzed by the esterases in the plasma, yielding the parent molecule [3].
What about those other differences between anabolic steroids?
In essence, the differences in biological effects of the various anabolic steroids are the result of their differences in chemical structure. I will go over some of these structural differences and their consequences.
The C-3-ketone group
If you look above at the picture of the steroid nucleus, you can see the third carbon atom in the bottom left. It’s part of the A ring and is extremely important in binding the androgen receptor (AR). The C-3-ketone group, as can be found in testosterone, looks like this (colored in green):
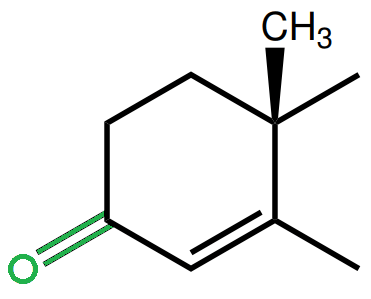
It’s an oxygen atom attached to the third carbon atom by a double bond. The importance of this group for receptor binding was highlighted by experiments by Ojasoo et al. [4]. In their paper “Towards the mapping of the progesterone and androgen receptors” they determined the relative binding affinities of 33 steroid ligands for the androgen receptor. This included the extremely potent androgen R1881 (methyltrienolone) and its “chemical brother” R3773, which lacks the ketone group at carbon 3. While R1881 demonstrated very high binding affinity for the androgen and progesterone receptor, and even some notable binding affinity for the glucocorticoid receptor, R3773 showed complete lack of any binding affinity as a result of this single structural modification.
Indeed, we see something similar going on when the C-3-ketone group gets reduced. That is, the oxygen atom attached with a double bond becomes a hydroxyl group (-OH). Take dihydrotestosterone (DHT) for example. It can be reduced on carbon 3 to yield either 3α- or 3β-androstanediol. This reaction is catalyzed by 3α-HSD or 3β-HSD, respectively. Both of these reduced metabolites of DHT have tremendously lower affinity for the androgen receptor [4]. This alteration looks as follows when looking at the chemical structure:
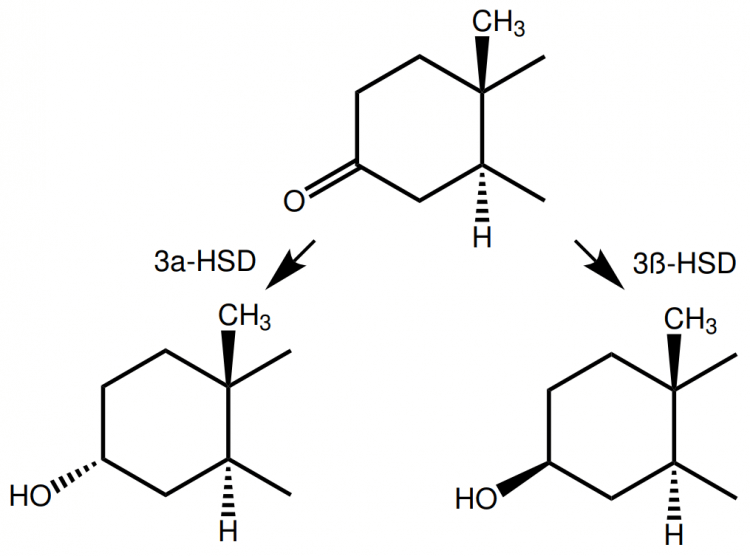
Now I also have a good excuse to explain the difference between something that is α-oriented and something that is β-oriented. Something that is α-oriented is illustrated using a dashed wedge, whereas something that is β-oriented is illustrated using a solid wedge. You should view a dashed wedge as going into the paper, whereas a solid wedge is coming out of the paper. The real molecule that is represented by our 2-dimensional drawings is, actually, 3-dimensional. Hence by drawing it with dashed and solid wedges, we’re able to incorporate the 3-dimensional structure of a molecule into our 2-dimensional drawings.
Either way, this reduction significantly lowers its binding affinity for the androgen receptor and this is a process that’s actively happening in skeletal muscle. Indeed, intravenous injection of labeled DHT showed a large breakdown (73.8 – 81.9 %) within 20 to 60 minutes in skeletal muscle compared to testosterone (20.2 – 30.5 %) [5]. The major DHT metabolite was 3α-androstanediol. This is the likely explanation of why, despite its several-fold higher binding affinity for the AR, DHT shows similar muscle-building effects as testosterone with equimolar exposure.
Back to where we were: the C-3-ketone group. What makes it so special for binding? Feel free to skip this and the next paragraphs, as I’m gonna grind you through a bit of organic chemistry. The reason is because the oxygen atom of the keto group has a lone pair of electrons. As such, it can function as a hydrogen bond acceptor to form a hydrogen bond (H-bond). And there appear to be two residues of the androgen receptor that are located in its ligand-binding domain (LBD) that do so [6]. The LBD is the part of the androgen receptor that’s directly interacting with a ligand, such as testosterone. It’s the site of the androgen receptor to which anabolic steroids bind. Much like how a key can fit into a lock, a ligand fits into the LBD (or ligand-binding pocket I should say) of a receptor. Without the C-3-ketone group, the ligand simply doesn’t fit as well, as the H-bonds that keep the ligand in the right place of the LBD, cannot be formed. As a rule, lack of a C-3-ketone group makes a steroid a more shitty key for its lock.
But Peter, what about Winstrol (stanozolol)? Indeed, stanozolol is an odd ball as it has a pyrazole ring fused to the A ring. For reference, here’s how that looks like and, as you can see, there’s of course no ketone group at carbon 3:
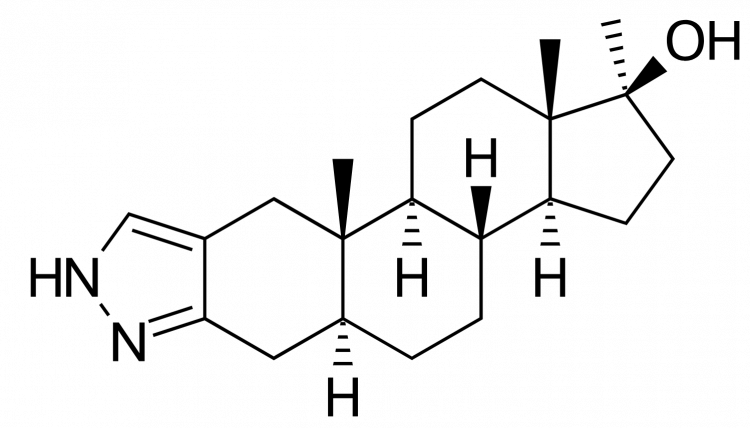
However, it’s assumed that the second nitrogen atom is forming the hydrogen bond in this case [6]. (Nevertheless, it’s affinity is still significantly lower.)
The C-17β-hydroxyl group and C-17α-alkylation
Whereas the C-3-ketone group can serve as a hydrogen bond acceptor, the C-17β-hydroxyl group can serve as a hydrogen bond donor. Two residues in the LBD of the AR can function as its corresponding hydrogen bond acceptor to form a H-bond [6]. And whereas the C-3-ketone group (=O) might be reduced to a hydroxyl group (-OH), the C-17 hydroxyl group (-OH) might be oxidized to form a ketone group (=O). This reaction is catalyzed by a class of enzymes named 17β-HSD [8]. This reaction is reversible. Several enzymes of this class exist, each chiefly catalyzing a unidirectional reaction. That is, some catalyze the oxidation reaction, forming the ketone group (=O), whereas others catalyze the reduction reaction, forming the hydroxyl group (-OH).
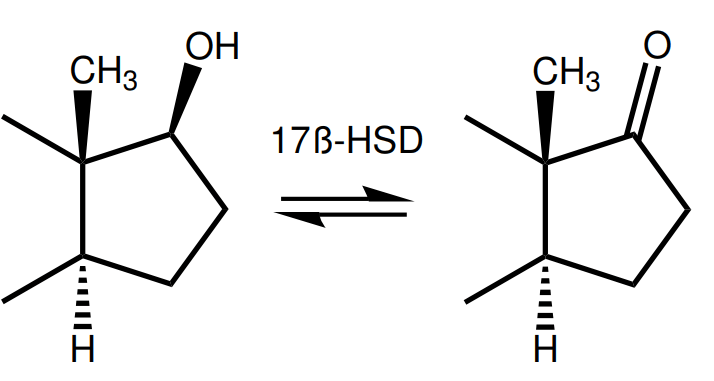
Just as with reduction or elimination of the C-3-ketone group, oxidation or elimination of the C-17β-hydroxyl group reduces the binding affinity for the AR.
The oxidation reaction, extensively, takes place in the liver. This is most likely one of the primary reasons why anabolic steroids have such low oral bioavailability. For example, only 3.66 % of orally administered testosterone became systematically available in one experiment [9]. Once it passes the liver, already a substantial amount will be metabolized.
In order to counteract this, an C-17α-alkyl group might be attached (commonly a methyl group). It is thought that the addition of this group hinders this oxidation through steric hindrance. Or, in other words, the methyl group gets in the way of the enzyme to properly act on the molecule and catalyze the reaction.
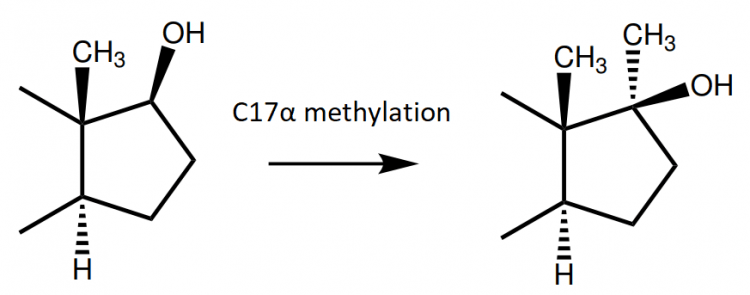
Also, as a result, steroids which possess this methyl group have a prolonged half-life of several hours compared to their nonmethylated counterparts. (Note that the long half-life of esterified anabolic steroids isn’t the result of slowed metabolism, but delayed diffusion out of the oil depot.) This resistance against hepatic metabolism is likely also the reason why they’re inherently hepatotoxic [10]. The C-17α methyl group, in combination with several other structural modifications, also enables the steroid to strongly bind to several nuclear receptors. This is very evident with the C-17α-methylated compound methyltrienolone, which demonstrates very strong affinity for the androgen receptor [4], but also has significant affinity for the glucocorticoid receptor [4], progesterone receptor [4, 11], and mineralocorticoid receptor [12].
The C-4,5 double bond
The difference between testosterone and dihydrotestosterone (DHT) is the addition of an α-oriented hydrogen atom on carbon 5 and, consequently, the conversion of the double bond between carbon 4 and 5 to a single bond. This is a reaction that is catalyzed by the class of enzymes known as 5α-reductase. Similarly, 5β-reductase adds a β-oriented hydrogen atom on carbon 5.
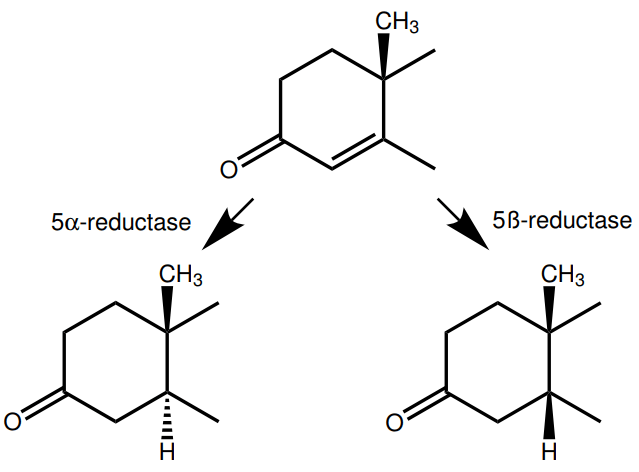
C-5α reduction of a compound can affect the binding affinity for the androgen receptor for the resulting metabolite. In the case of testosterone, it increases the binding affinity. (The C-5α reduction of testosterone yields DHT, which has significantly higher affinity.) The result of this is that testosterone’s effects can get amplified in tissues expressing 5α-reductase, such as the scalp. This is why an important treatment modality for androgenetic alopecia (male-pattern baldness) inhibits this enzyme (Finasteride, a 5α-reductase inhibitor).
However, when nandrolone undergoes C-5α-reduction, it forms dihydronandrolone (DHN): a compound with considerably less affinity for the androgen receptor [13]. Resultingly, nandrolone’s actions can be dampened in tissues expressing 5α-reductase. In general, C-5α-reduction only appears relevant for testosterone and nandrolone, as most commonly used AAS are either already C-5α-reduced, or don’t undergo significant C-5α-reduction in the body.
| Compound | C-5α-reduced in the body? |
| Testosterone | Yes, to a more potent androgen (DHT) |
| Nandrolone | Yes, to a less potent androgen (DHN) [13] |
| Boldenone | No, not significantly C-5α-reduced in the body [14] |
| Trenbolone | No, not significantly C-5α-reduced in the body [14] |
| Methandienone | No, not significantly C-5α-reduced in the body [14] |
| Turinabol | No, not significantly C-5α-reduced in the body [14] |
| Fluoxymesterone | No, not significantly C-5α-reduced in the body [14] |
| Methenolone | No, already C-5α-reduced |
| Drostanolone | No, already C-5α-reduced |
| Stanozolol | No, already C-5α-reduced |
| Oxandrolone | No, already C-5α-reduced |
| Oxymetholone | No, already C-5α-reduced |
One of the functions of 5β-reductase in the human body is to deactivate testosterone and turn it into metabolites with hardly any affinity for the androgen receptor. Together with 5α-reductase, these enzymes catalyze the what is thought to be the initial and rate-limiting step in the metabolism of androgens with a C-3-ketone group and a double bond between carbons 4 and 5, i.e. 3-keto-4-ene androgens such as testosterone [14]. 5β-reductase is predominantly expressed in the liver [15]. Finally, whereas DHT has a very high affinity for the androgen receptor, its C-5β-reduced brother hardly has affinity for it [4].
Removal of C-19 (“19-nor”)
If you remove the C-19-methyl group of the testosterone molecule, you’ll have nandrolone (also known as 19-nortestosterone). One of the consequences of this, as described above, is that subsequent C-5α-reduction yields a less potent androgen (DHN) instead of a more potent androgen, as would be the case with testosterone (namely DHT). Additionally, the removal of this methyl group appears to affect the rate of aromatization. A study in human placental microsomes observed a relative activity of aromatization that was only 20 % of that of testosterone [16]. Suppression of gonadotropins relies partly on the aromatization of an androgen in estrogen to suppress at the level of the pituitary [17]. Indeed, administration of 200 mg nandrolone hexyloxyphenylprionate every 3 weeks for several months was unable to completely suppress gonadotropins and testosterone [18]. This is in line with the premise that relatively little aromatization of nandrolone occurs. Finally, this chemical alteration gives the compound significant affinity for the progesterone receptor [4, 11].
Another well-known anabolic steroid that lacks the C-19-methyl group is trenbolone. Similar to nandrolone, it also demonstrates significant affinity for the progesterone receptor. However, in contrast with nandrolone, no C-5α/β-reduced metabolites have been detected for the compound [14]. As such, the two other chemical alterations in trenbolone versus nandrolone appear to prevent this (the addition of double bonds in the B and C rings). This also appears to prevent aromatization of the A ring, demonstrating very low estrogenic activity even at extremely high concentrations [19].
References
- Von Eckardstein, Sigrid, and Eberhard Nieschlag. “Treatment of male hypogonadism with testosterone undecanoate injected at extended intervals of 12 weeks: a phase II study.” Journal of andrology 23.3 (2002): 419-425.
- Kalicharan, Raween Wikesh. New Insights into Drug Absorption from Oil Depots. Diss. Utrecht University, 2017.
- Van der Vies, J. “Implications of basic pharmacology in the therapy with esters of nandrolone.” European Journal of Endocrinology 110.3_Suppla (1985): S38-S44.
- Ojasoo, T., et al. “Towards the mapping of the progesterone and androgen receptors.” Journal of steroid biochemistry 27.1-3 (1987): 255-269.
- Becker, H., et al. “In vivo uptake and metabolism of 3H-testosterone and 3H-5α-dihydrotestosterone by human benign prostatic hypertrophy.” European Journal of Endocrinology 71.3 (1972): 589-599.
- Pereira de Jésus‐Tran, Karine, et al. “Comparison of crystal structures of human androgen receptor ligand‐binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity.” Protein Science 15.5 (2006): 987-999.
- Feldkoren, Boris I., and Stefan Andersson. “Anabolic-androgenic steroid interaction with rat androgen receptor in vivo and in vitro: a comparative study.” The Journal of steroid biochemistry and molecular biology 94.5 (2005): 481-487.
- Luu-The, Van. “Analysis and characteristics of multiple types of human 17β-hydroxysteroid dehydrogenase.” The Journal of steroid biochemistry and molecular biology 76.1-5 (2001): 143-151.
- Täuber, U., et al. “Absolute bioavailability of testosterone after oral administration of testosterone-undecanoate and testosterone.” European journal of drug metabolism and pharmacokinetics 11.2 (1986): 145-149.
- Bond, Peter, William Llewellyn, and Peter Van Mol. “Anabolic androgenic steroid-induced hepatotoxicity.” Medical Hypotheses 93 (2016): 150-153.
- Houtman, Corine J., et al. “Detection of anabolic androgenic steroid abuse in doping control using mammalian reporter gene bioassays.” Analytica chimica acta 637.1-2 (2009): 247-258.
- Takeda, Armelle-Natsuo, et al. “The synthetic androgen methyltrienolone (r1881) acts as a potent antagonist of the mineralocorticoid receptor.” Molecular pharmacology 71.2 (2007): 473-482.
- Bergink, E. W., et al. “Comparison of the receptor binding properties of nandrolone and testosterone under in vitro and in vivo conditions.” Journal of steroid biochemistry 22.6 (1985): 831-836.
- Schänzer, Wilhelm. “Metabolism of anabolic androgenic steroids.” Clinical chemistry 42.7 (1996): 1001-1020.
- Chen, Mo, and Trevor M. Penning. “5β-Reduced steroids and human Δ4-3-ketosteroid 5β-reductase (AKR1D1).” Steroids 83 (2014): 17-26.
- Ryan, Kenneth J. “Biological aromatization of steroids.” Journal of Biological Chemistry 234.2 (1959): 268-272.
- Pitteloud, Nelly, et al. “Inhibition of luteinizing hormone secretion by testosterone in men requires aromatization for its pituitary but not its hypothalamic effects: evidence from the tandem study of normal and gonadotropin-releasing hormone-deficient men.” The Journal of Clinical Endocrinology & Metabolism 93.3 (2008): 784-791.
- Behre, H. M., et al. “Suppression of spermatogenesis to azoospermia by combined administration of GnRH antagonist and 19-nortestosterone cannot be maintained by this non-aromatizable androgen alone.” Human Reproduction 16.12 (2001): 2570-2577.
- Le Guevel, Rémy, and Farzad Pakdel. “Assessment of oestrogenic potency of chemicals used as growth promoter by in-vitro methods.” Human reproduction 16.5 (2001): 1030-1036.
Image by Arek Socha from Pixabay
About the author

Peter Bond is a scientific author with publications on anabolic steroids, the regulation of an important molecular pathway of muscle growth (mTORC1), and the dietary supplement phosphatidic acid. He is the author of several books in Dutch and English, including Book on Steroids and Bond's Dietary Supplements.
Leave a Reply
You must be logged in to post a comment.