Table of Contents
- Bro-science myths
- “Insulin supports exercise-induced fat oxidation by blunting hyperglycemia”
- “Insulin protects the pancreas by giving the pancreatic islet β-cell a break”
- “Insulin enhances dieting by directing energy to muscle protein synthesis via glycogen synthesis and storage rather than to fat storage in an energy deficit because fat storage is impossible in a deficit”
Introduction and Purpose
This article is intended to provide a comprehensive understanding on insulin actions for the bodybuilder and biochemist alike. Its primary purpose is to counter a narrative, currently promulgated by the scientific community’s gatekeepers, that insulin is (supposedly) not muscle protein anabolic, but rather merely promotes increased glycogen storage transiently.
This is laughably untrue, as the reader will learn, if not yet convinced by the potent effects readily apparent in insulin-using bodybuilders, typified by the so-called “mass monsters.”
It is this author’s view that this narrative is akin to the pre-Bhasin (2001) view of anabolic-androgenic steroids as not possessing muscle anabolic effects.
At present, the scientific community will have you believe that exogenous insulin is not particularly anabolic in human skeletal muscle.
These excerpts from my notes on insulin will strongly contend with this institutional bias (analogous to pre-Bhasin view on androgens).
The institutional blind spot arises from a lack of study on the use of exogenous insulin with:
- supraphysiological dosing
- administered systemically
- hyperaminoacidemia
- in conjunction with supraphysiological rhGH (and, more often than not, anabolic-androgenic steroids)
The secondary purpose of this article is to address, and dispel where necessary, bro-science claims, that include the claims that:
- “Insulin supports exercise-induced fat oxidation by blunting hyperglycemia”
- “Insulin protects the pancreas by giving the pancreatic islet β-cell a break”
- “Insulin enhances dieting by directing energy to muscle protein synthesis via glycogen synthesis and storage rather than to fat storage in an energy deficit because fat storage is impossible in a deficit”
Terminology and Definitions
AA: amino acid
AAS: anabolic-androgenic steroids
AV: ateriovenous
CHO: carbohydrate
CV: cardiovascular
FAT: lipid and/or fat
MPB: muscle protein breakdown
MPS: muscle protein synthesis
PRO: protein
RhGH: recombinant human growth hormone (Somatropin)
RT: resistance training
Slin: recombinant human insulin (rhI)
Insulin actions
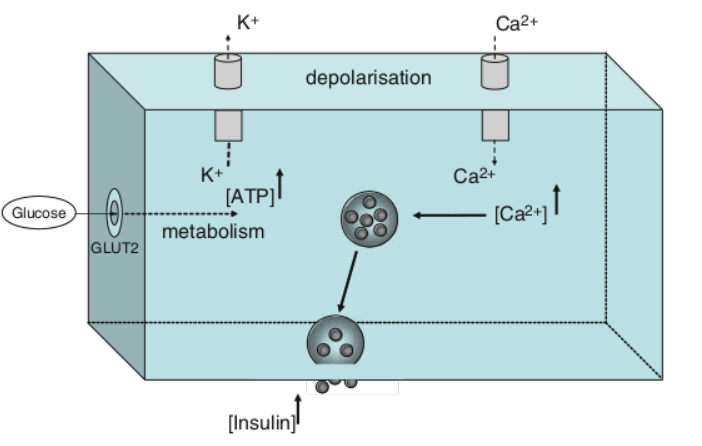
The primary effect of insulin in skeletal muscle is on the electrochemical transmembrane gradient, where it induces hyperpolarization in the muscle cells by directly activating the Na⁺-K⁺-ATPase pump (46). [2]. Insulin increases the number of Na+/K+-ATPase pumps in the membrane as well, leading to an intracellular shift of potassium, causing hypokalemia (low K/potassium) in the extracellular space of the blood (serum). [1].
Insulin stimulates gene expression of MHCα (slow isoform) [16] in skeletal muscle and albumin in the liver (Dillman, 1988). [6].
Conversely, insulin seems to stimulate both type I & II fibers equally [21]:
This paper investigated whether proteins involved in glucose metabolism [we care most about Akt phosphorylation & glycogen storage capacity] are expressed and/or regulated by insulin in a fiber type-specific manner in human skeletal muscle.
The relevant findings and portion of the discussion:
Higher protein levels of IRβ (+16%), hexokinase II [commits glycogen to the cell] (+470%), GLUT4 [transports glucose into the cell] (+29%), and electron transport chain complex II [mitochondrial/Kreb’s cycle] (+35%) was found in type I vs type II fibers…
Akt, mTOR: Akt2 protein content was lower (-27%) in type I vs type II fibers…the average increases under insulin stimulation of [Akt phosphorylation] (hypertrophy) were 5.8 and 3.5-fold in type I fibers and 6.1 & 3.7-fold in type II fibers, respectively…. the relative response to insulin was similar between fiber types.
From Discussion:
Human type I fibers have a greater abundance to transport (+29% GLUT4), phosphorylate (+470% HKII) and oxidize (+35% ETC complex II and +34% pyruvate dehydrogenase complex) glucose & to synthesize glycogen (+35%) compared to type II fibers.
Type I fibers possess greater glycogen storage capacity.
The apparent fiber-type differences in insulin-stimulated phosphorylation of Akt, NDRG1 (downstream of mTOR [these are our hypertrophy pathways])… were eliminated when adjusted for…protein abundance. These findings suggest a similar sensitivity of type I and type II muscle fibers for regulation by insulin of the proteins investigated. [21]
This data [21] supports a view that, rather than preferentially promoting muscle protein anabolism in the slow-twitch fibers, both type I & II muscle fibers are equally responsive to the protein anabolic effects of exogenous insulin.
Hyperinsulinemia in normal subjects acutely decreases plasma concentrations of amino acids as a consequence of net protein deposition. [6].
Once secreted from the pancreatic beta-cells, insulin circulates in the blood stream with an approximate half-life of approximately 12 min. Numerous tissues and organs express the insulin receptor and various actions are initiated (Sonksen 2001), some of which are of general importance as well as particular interest for elite sports.
The central effect of insulin in concert with other hormones such as glucagon or somatostatin is the control of blood glucose levels. The secretion of insulin in response to elevated glucose concentrations (e.g., postprandial) inhibits hepatic glucose production, which subsequently lowers blood glucose levels. In contrast, glucagon, which is composed of 29 amino acid residues, is a counterregulatory hormone of insulin. It raises plasma glucose levels in response to insulin-induced hypoglycemia and plays an important role in glucose homeostasis by increasing gluconeogenesis and decreasing glycolysis. The third pancreatic hormone relevant for the control of blood glucose levels is somatostatin, a 14-residue peptide that exerts inhibitory effects on the secretion of insulin but not on its biosynthesis. The entire mechanism of its action has not yet been clarified but studies suggest a paracrine effect that inhibits the exocytosis of insulin from cells adjacent to somatostatin-producing D-cells (Reichlin 1983).
However, the effects of insulin on the entire organism are manifold and complex. Insulin causes for instance the translocation of GLUT-4 (the glucose transporter predominantly found in skeletal muscle and adipose tissue) from intra-cellular vesicles to the cell membrane and, thus, increases the rate of glucose entry for a given concentration into the target tissue. An excess of glucose transferred into cells subsequently stimulates glycogen formation (Halse et al. 2001; Yeaman et al. 2001), a fact that is of considerable interest in endurance sports where the amount of glycogen stored in muscle cells can influence athletic performance. Moreover, the (muscle) protein metabolism is significantly influenced by the chalonic [restraining, inhibitory] (Rooyackers and Nair 1997; Sonksen 2001) as well as the stimulating properties of insulin (Biolo et al. 1995; Biolo and Wolfe 1993; Tipton and Wolfe 2001; Wolfe 2000, 2005). Due to the anti-catabolic effect of insulin, protein breakdown is significantly reduced and allows the preservation of contractile muscle elements. In addition, synthesis-stimulating effects were observed in numerous studies that demonstrated the anabolic properties of insulin, utilizing for instance stable-isotope labeled analogs. The anabolism, however, was strongly dependent on amino acid availability (Fujita et al.2006; Garlick and Grant 1988; Zhang et al.1999). [1]
See Intracellular AA availability for a more in-depth explanation on the importance of amino acid availability for insulin muscle anabolic effects.
Tissue Effects on Carbohydrate, Lipid, and Protein Metabolism
The major effects of insulin on tissues are:
(1) Carbohydrate metabolism:
(a) It increases the rate of transport of glucose across the cell membrane in adipose tissue and muscle
(b) it increases the rate of glycolysis in muscle and adipose tissue
(c) it stimulates the rate of glycogen synthesis in a number of tissues, including adipose tissue, muscle, and liver. It also decreases the rate of glycogen breakdown in muscle and liver
(d) it inhibits the rate of glycogenolysis and gluconeogenesis in the liver.
(2) Lipid metabolism:
(a) It decreases the rate of lipolysis in adipose tissue and hence lowers the plasma fatty acid level
(b) it stimulates fatty acid and triacylglycerol synthesis in tissues, although only to a minor extent in humans
(c) it increases the rate of very-low-density lipoprotein formation in the liver
(d) it increases the uptake of triglyceride from the blood into adipose tissue and muscle
(e) it decreases the rate of fatty acid oxidation in muscle and liver
(f) it increases the rate of cholesterol synthesis in liver.
(3) Protein metabolism:
(a) It increases the rate of transport of some amino acids into tissues
(b) it increases the rate of protein synthesis in muscle, adipose tissue, liver, and other tissues
(c) it decreases the rate of protein degradation in muscle (and perhaps other tissues)
(d) it decreases the rate of urea formation.–These insulin effects serve to encourage the synthesis of carbohydrate, fat and protein. [26]
Mechanisms in skeletal muscle hypertrophy
An anabolic hormone may induce skeletal muscle hypertrophy by a) increased MPS or b) reduced proteolysis. Further, c) transmembrane transport of AAs (AA trafficking) may be under hormonal control.
Insulin induces skeletal muscle hypertrophy by all three of these aspects of muscle protein metabolism.
(a) profoundly (“the fractional synthesis rate of muscle protein was ~65% higher (P = 0.02) during insulin infusion”) [2]
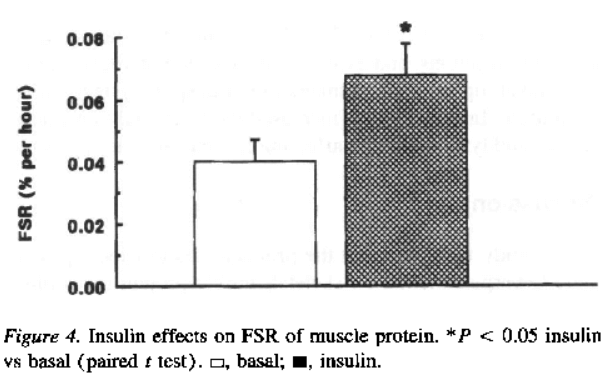
(b) modestly (in nonmyofibrillar skeletal muscle protein)
(c) by influencing the activity of at least four distinct AA transport systems (not a primary mediator of insulin’s action on skeletal muscle hypertrophy):
- A, primarily: transports AAs with short polar side chains such as alanine and glycine; not rate-limiting for protein synthesis
- ASC
- Nᵐ, substantially
- Xc [2]
…in severely burned patients…insulin infusion (10 – 12 IU / hr, 70 kg patient) ↑MPS without affecting MPB (28). [24]. No change in AA uptake into skeletal muscle, suggesting the muscle anabolic effects resulted from more efficient reutilization of AAs generated by proteolysis (28). [24].
IGF-I
Additionally, insulin ↑IGF-I bioavailability.
Insulin may enhance the bioavailability of IGF-I by inhibiting production of IGFBP-1 & increasing IGFBP-3-proteolytic activity, which reduces the affinity of IGF-I for IGFBP-3 (29). [24].
* IGF-I (free) ↑AA & glucose uptake & ↓protein breakdown (29). [24].
Muscle Protein Synthesis
Insulin’s skeletal muscle protein synthesis is modulated by insulin-induced changes in:
(a) muscle blood flow, and
(b) AA availability. [10]. That is, insulin must be considered in the context of (AA concentration x flow [i.e., increased delivery of AA to muscle]). [8].
+ This presents a counterargument against Wolfe’s view that insulin must stimulate AA reincorporation from MPB back into intracellular MPS… Wolfe argued that without this, insulin must increase AA availability by some other means (than reincorporation) such as by ingestion or infusion (which causes hypoaminoacidemia). [8].
Specifically, all studies in which MPS had been stimulated by insulin also had an increased AA delivery to the muscle tissue (amino acid concentration x blood flow)…
The differences in AA delivery were mainly due to differences in AA concentrations, which, in turn, were determined by the modality of insulin infusion (systemic or local) and/or the concomitant infusion of exogenous AAs. This is because systemic insulin infusion decreases blood AA concentrations unless AAs are replaced by exogenous infusion.
Conversely, local insulin infusion in a leg or a forearm allows for the exposure of the muscle tissue to relatively high insulin levels while avoiding a major reduction in blood AA concentration. [10].
Insulin > IGF-I in human skeletal muscle anabolism [25]
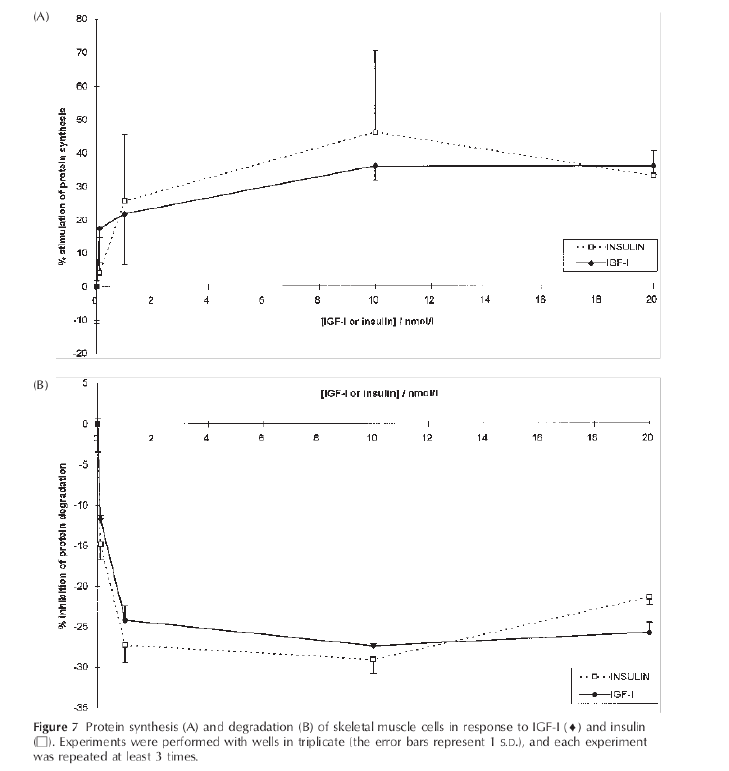
Insulin directs MPS by direct activation of the PI3K/Akt/mTORC1 pathway and due to its increased muscle blood flow via vasoactive properties. [20].
Purported interference effect of rhGH
Fryburg, et al., [13] proposes that rhGH & exogenous insulin work at cross-purposes, that growth is a consequence of muscle protein turnover rather than hypo/hyperglycemic hormonal action. Coadministration of endogenous levels of insulin and rhGH merely attenuates the protein anabolic effects of GH, and is not additive.
In practice, this has been thoroughly dismissed. Mocked, even. And this is because of the importance of Intracellular AA availability:
Intracellular AA availability
The first step in protein synthesis occurs in the nucleus of the cells and involves the transcription of a specific gene into mRNA molecules. mRNAs are transported into the cytosol, where they associate with ribosomes for the translationof the base sequence into an AA sequence. The translation process can be divided into three phases: initiation, elongation, and termination of the peptide chain. Many ribosomes can associate with mRNA molecules, forming polyribosomes. A polyribosome can synthesize several peptide chains from a single mRNA. Cytoplasmic free AAs are not directly utilized for protein synthesis. The translation process involves the binding of AAs to specific transfer RNA (tRNA) molecules, forming aminoacyl-tRNAs. The aminoacyl-tRNAs constitute very small pools that are turning over rapidly. [6].
Insulin has been shown to stimulate both the transcription and translation processes for specific proteins (Kimball and Jefferson, 1988). Gene expression (as reflected by mRNA levels) for many proteins has been shown to be stimulated by insulin, including albumin in liver and MHC α in skeletal muscle (Dillman, 1988). The mRNA content reflects the ultimate potential for protein synthesis in the (tissue) cell, but may not be directly related to the rate of translation and thus synthesis of protein. Insulin also enhances the translation process in skeletal muscle by stimulating peptide chain initiation (Jefferson et al, 1974; Fulks and Goldberg, 1975). Thus from the molecular basis it would be expected that in vivo insulin would generally increase the overall capacity for protein synthesis (mRNA content) and stimulate the translation and production of specific proteins, such as muscle proteins. [6].
There are two mechanisms by which increased intracellular AA may stimulate muscle protein synthesis. First, a mass-action effect from the supplied exogenous AAs; tRNA necessary for translation of AAs to proteins is available in muscle in amounts that exceed the available AA pool. Accordingly, increasing the AA availability will drive MPS simply by charging more tRNA. [7].
It is also possible that certain AAs, either individual AAs or groups of AAs (e.g., BCAAs), may signal initiation of the translation process (ribosomal). Rennie et al (56) demonstrated that single essential AAs given in a flooding dose [ie, filling the AV space as well as the intracellular space] stimulate MPS, whereas nonessential AA have no such efect. Moreover, we have recently shown that it is unnecessary to include nonessential AAs in a solution that simulates muscle anabolism in resting volunteers (63). [7].
These studies are consistent with the hypothesis that one or more of the essential AAs may act as a signal for initiation of MPS. However, stimulation of protein synthesis through mass action cannot be ruled out based solely on this evidence. It is possible that sufficient nonessential AAs are present and that the addition of large amounts of essential AAs provides enough tRNA charging to drive protein synthesis. [7].
Wolfe view: Insulin apparently enhances the efficiency of the reincorporation of AAs resulting from protein breakdown (intracellular) back into newly synthesized protein. [2] [8].
AA availability as a signal to activate translation
Ubiquitin-proteosome system (predominant MPB system in resting muscle)
Eukaryotic initiation factors (eIF), in particular eIF4E, are important components of the control of the initiation of translation. When eIF4E forms a complex with eIF4G, initiation of translation is promoted. Binding of eIF4E to eIF4G is normally prevented because eIF4E is bound to a binding protein, eIF4E-BP1, phosphorylation of which releases eIF4E. Provision of AAs enchances the phosphorylation thereof, thereby allowing protein synthesis to proceed (64). That is, the formation of the active eIF4E-eIF4G complex is increased in response to protein feeding (64, 75). [7].
AA availability as a chalonic to muscle proteolysis
The intracellular AA pool seems to likewise follow a mass-action effect to stave off muscle protein breakdown.
An increase in intracellular AAs only inhibits MPB under certain conditions. For example, hyperaminoacidemia at rest has little or no effect on MPB. Yet, when AAs are given post-RT, when MPB would normally be elevated there is no increase in MPB. This conditional inhibition of MPB is consistent with the notion that this relationship between AA availability and reduced MPB occurs mainly when the intracellular available AA pool becomes depleted. For example, if the initial response to exercise is increased MPS, then the AA pool is reduced, leading to MPB to maintain the intracellular AA pool. [7].
Alternatively, because there are different pathways of MPB, it is possible that following exercise, the lysosomal pathway becomes predominant in mediating the acceleration of MPB, and this pathway may be responsive to AA availability [and exogenous insulin!]. [7].
…The only likely effect of carbohydrates and fats on net protein balance is by hormonal influence as substrates [e.g., CHO influence on insulin secretion]. [7].
Proteolysis
During adequate AA supply, the most important degradative system in muscle is the ATP-independent ubiquitin system. [2]. This system is not sensitive to insulin [2]. Insulin apparently plays a role only in the regulation of the lysosome activity [2]. Lysosomes (organelles) are not involved in the myofibrillar protein degradation in normal conditions, but only in the presence of low insulin levels or decreased AA availability. [2].
The whole-body insulin-induced reduction in skeletal muscle protein breakdown occurs in nonmyofibrillar proteins (mostly liver). [5].
AA transmembrane transport
Alanine +48%, Lysine +75%, Leucine +22%. [2].
Alanine is a major substrate of system A, a sodium-dependent system that maintains steep transmembrane gradients of AA concentrations, and is regulated by insulin [2] . Further, alanine synthesis in the muscle cell increases due to the insulin-mediated increase of glucose uptake and intracellular production of pyruvate in muscle. [2].
Lysine is a cationic AA that is transported by the sodium-independent system y⁺ that is strongly influenced by the electrochemical potential of cell membrance. Since insulin induces hyperpolarization in the skeletal muscle cells by directly activating the Na⁺-K⁺-ATPae pump, acceleration of lysine transport may be secondary to the primary insulin effect on the electrochemical transmembrane gradient. [2].
The branched-chain (leucine, isoleucine, and valine) and the aromatic (phenylalanine and tyrosine) AAs are preferably transported through system L. This sodium-independent system is unable to generate high transmembrane gradients for its substrates. It has been shown that the kinetic characteristics of system L are not influenced by insulin. [2].
Insulin action on leucine is particularly modulated by increased muscle blood flow! Leucine is subject to oxidation and transamination that must be subtracted from the rate of disappearance in muscle to obtain protein synthesis rates with the arteriovenous balance technique. [5].
Antilipolytic
Insulin is antilipolytic leading to attenuated fat availability and oxidation during exercise (DeMarco et al., 1999; Stevenson et al. 2005).
Insulin is the primary inactivator of hormone sensitive lipase (HSL) activity, down-regulating mobilization of triglycerides within adipocytes. [28].
* Even fasting insulin levels (1.4 – 14.0 µIU/mL or 9.7 – 97.2 pmol/L) are sufficient to inactivate HSL by ~50%. Small increases in insulin (from either PRO or CHO intake) inactivate HSL further.
Somatostatin
Pancreatic islet cells:
- α: secrete glucagon
- β: secrete insulin
- δ: secrete somatostatin (primarily). [3].
Glucagon (catabolic) and insulin interact via a classical negative-feedback loop (glucagon stimulates somatostatin secretion; somatostatin inhibits secretion of glucagon). Glucagon is elevated in the fasted state in order to signal to the liver to break down stored glycogen.
Exogenous insulin administration does not affect somatostatin secretion. [3]. Conversely, insulin insufficiency can cause hypersecretion of somatostatin.
Mechanisms of action (primary): blocks Ca-mediated intracellular activation; blocks K⁺ outflux from pancreatic cells, which in turn ↓ Ca⁺ intracellular accumulation, which in turn disrupts glucose-induced stimulus-secretion coupling. [3].
Glucose-Insulin interplay
The two phases of insulin response to glucose administration (immediate & prolonged) are differentially influenced by somatostatin. The initial phase is extremely sensitive to somatostatin inhibition, whereas the latter effects require much more hormone. [3].
Carbohydrates (and AAs/smaller parts of proteins) are broken down to blood glucose. In the postprandial state, insulin is secreted by pancreatic β cells, which transports glucose from the blood to the cells of the body (e.g., skeletal muscle), where it is used for energy. [3].
Blood glucose levels
In a healthy state, the organism balances insulin and glucose homeostatically.
Normal blood glucose levels are:
- fasted: 70 – 88 mg/dL
- 2 hours post-prandial: ≤ 140 mg/dL
* <70 mg/dL is hypoglycemia
In response to meals with varying carbohydrate, insulin levels may rise to the range of approximately 300-800 pmol/L. [18].
Hyperglycemia is defined as > 6.6 mmol of glucose per L, fasting
Prolonged conditions of elevated glucose concentrations cause dysfunction of numerous cell types in the body, including beta cells, neurons, and the endothelium, via several pathway, including increased oxidative stress and activation of the sorbitol pathway (31-33). [18].
Glucometer measures
* Fasting blood glucose levels upon waking: 70 – 100 mg/dL or 3.9 – 5.5 mmol/L
* Blood glucose levels 2 hours after meal: 90 – 130 mg/dL or 5.0 – 7.2 mmol/L
* Fasting hyperglycemia: blood glucose > 130 mg/dL or 7.2 mmol/L after fasting for at least 8 h
* Postprandial hyperglycemia: blood glucose > 180 mg/dL or 9.9 mmol/L, 2 h after eating a meal containing carbohydrates. Healthy persons rarely have blood glucose > 140 mg/dL after consuming a meal unless the meal contained a large amount of processed carbohydrates (cereal, popcorn, cake, etc.)
* Fasting & postprandial hypoglycemia: blood glucose < 70 mg/dL or 3.9 mmol/L after fasting for at least 8 h as well as between meals
Insulin resistance
A state wherein the liver doesn’t recognize the body’s insulin and continues to produce glucose in inappropriately high quantities.
Marked by a hyperglycemic state caused by (on balance) insufficient pancreatic insulin secretion, such that insulin cannot “shuttle” enough glucose into the cell(s).
Regulation of insulin sensitivity occurs by several mechanisms – broadly, central mechanisms include GLP-1 & GIP receptors; peripheral mechanisms include transporter proteins, e.g. glucose transporters (e.g., GLUT-4 in skeletal muscle). HOMA-IR & QUICKI provide insight into systemic insulin sensitivity.
Ketoacidosis may result from these conditions, which is a condition caused by the release of FFAs from adipocytes due to insufficient insulin levels, whereby the liver is taxed with metabolizing FFAs, leading to a toxic systemic ketone level.
Type 2 diabetes mellitus (T2DM) is an insulin resistant form of diabetes, whereas type 1 diabetes is insulin sensitive. Type 1.5 diabetes is synonymous with LADA (latent autoimmune diabetes of adults), and is characterized by islet autoantibodies (e.g., glutamic acid decarboxylase antibody > 20 U/mL) and low C-peptide secretion. HbA1c is the primary indicator of a long-term hyperglycemia.
HOMA-IR
Fasting insulin * fasting glucose / 405
Optimal range: 0.5 – 1.4
Another form: Fasting serum insulin (μU/ml) * Fasting plasma glucose (mmol/liter) / 22.5
QUICKI
Quantitative Insulin Sensitivity Check Index (QUICKI): The inverse of the sum of the logarithms of the fasting insulin and fasting glucose:
1 / (log(fasting insulin μU/mL) + log(fasting glucose mg/dL))
IR-A, IR-B, & IGF-IR function, potency
Receptor function
Insulin and its recombinant formulations activate IR-A, IR-B, & IGF-IR.
The IR is a transmembrane receptor tyrosine kinase and exists in two isoforms: isoform A (IR-A) & isoform B (IR-B) [23] . The IR-A is expressed ubiquitously but is predominantly expressed in CNS, hematopoietic cells, and in cancer tissues (1-4) [23] . The IR-B is expressed predominantly in the liver, but is also substantially expressed in muscle & adipose tissue, the major target tissues for the metabolic effects of insulin (1-4) [23] . Both IR isoforms show great homology with the IGF-IR (4-6) [23] . The IGF-IR is found in most tissues and brings about mitogenic, pro-invasive, and anti-apoptotic effects (7). [23].
Epidemiological data has indicated a relationship between rhI, in particular insulin glargine (Lantus) & risk of cancer (8-10). [23].
Potency to activate
Insulin glargine activates the IGF-IR with significantly greater potency than its close cousin (and the short-acting rhI preparations tested): insulin detemir (Levemir), insulin aspart (NovoLog, NovoRapid), & insulin lispro (Humalog). [23].
EC₅₀(IR-B) [metabolic effects]: 1.8 ± 1.5 (insulin aspart; NovoLog, NovoRapid) > *; EC₅₀(IGF-IR) [mitogenic effects]: 13.9 ± 1.4 (insulin glargine; Lantus) >> * [23].
Insulin and morbidity
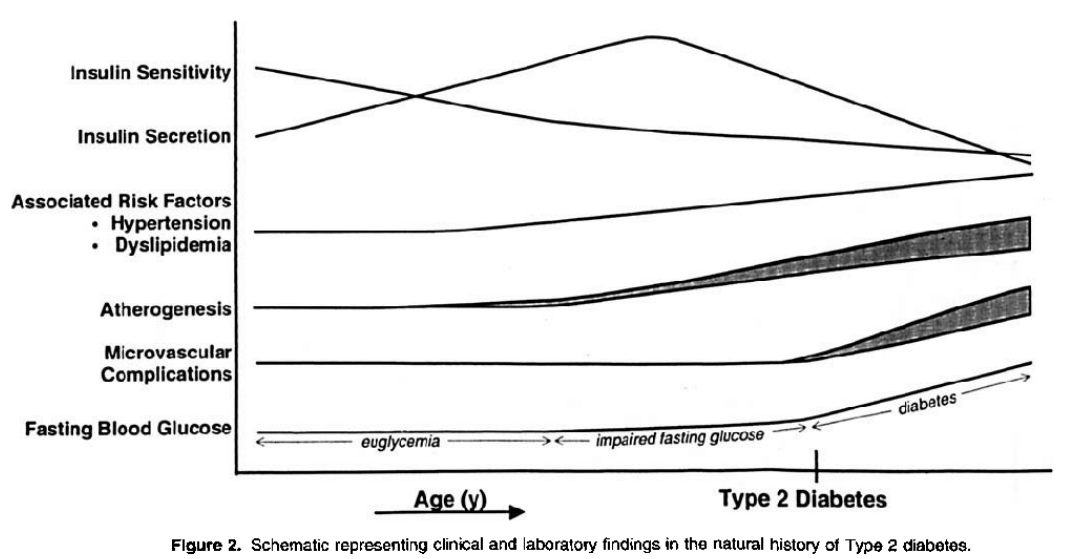
The association between elevated endogenous circulations of insulin levels is tied to insulin resistance, yet the causality of absolute or elevated insulin concentrations on the pathological course of progression to diabetes is not established.
Proposed natural history of T2D progression. [14].
Note the correlation between insulin secretion and insulin sensitivity.
Yet, there is no indication that insulin negatively feeds back at the pancreatic β cells to inhibit its pulsatile release.
Rather, the mechanisms of insulin resistance involve:
* impaired glucose transport via post-receptor defects in insulin signaling (i.e., GLUT-4 translocation; tyrosine kinase phosphorylation/signaling via the β-subunit of the IR) [14]:
Insulin resistance in most cases is believed to be manifest at the cellular level via post-receptor defects in insulin signalling. Despite promising findings in experimental animals with respect to a range of insulin signalling defects, their relevance to human insulin resistance is presently unclear. Possible mechanisms include down-regulation, deficiencies or genetic polymorphisms of tyrosine phosphorylation of the insulin receptor, IRS proteins or PIP-3 kinase, or may involve abnormalities of GLUT-4 function.[15].
Still, it is generally true that very high plasma insulin levels are a predictor of the development of diabetes [14]. There are, further, independent risk factors that the complicated use of AAS (and some contribution from rhGH) aggravate:
Independent risk factors for impaired insulin sensitivity
- central obesity
- elevated triglycerides; ↓HDL, ↑Apo B, ↓Apo A1 (dyslipidemia)
- endothelial dysfunction (altered arterial tone ⇒ atherosclerosis)
- atherosclerosis (factors: platelet adhesion, aggregation, thrombogenecity ⇒ inflammation)
- hypertension
- prothrombotic activity. [14].
Insulin therapy increases cardiovascular risk
Multiple analyses show a clear dose-dependency of increased CV morbidity and exogenous insulin (the hazard ratio increased 2- to 3- fold for CV death per 1 unit/kg/day)… and all-cause mortality, cardiovascular events, and cancer… and a proportionality between duration of insulin use and serious ischemic cardiac events: CV risk doubled in the first few months of insulin use, and tripled by 6 to 12 months. [19].
This increased risk seems to be ameliorated with the use of insulin glargine (a long-acting “basal insulin”). [22]. Thus, the increased cardiovascular risk is likely inherent to short-/rapid- acting rhI preparations due to hypoglycemia (often undetected and/or insufficiently treated), or perhaps increased IR activity/downstream phosphorylation (related most plausible to IR-A potency).
Insulin increases VLDL synthesis in liver
Insulin promotes synthesis of the very low density lipoprotein particles that promote atherosclerosis. [26].
Exogenous insulin-induced insulin resistance
“Insulin toxicity”
When cells are exposed to continuously elevated insulin levels, there is a partial downregulation of insulin signaling. The resuling “insulin resistance” (insulin toxicity, herein) is not primarily due to less IR expression on the cell surface but due to impaired insulin signal transduction due to receptor dysfunction. [18]. In response to prolonged hyperinsulinemia, there is diminished autophosphorylation of the IR, and subsequent PI3K-AKT signaling is affected (34, 35). [18]. Consequently there is attenuated GLUT-4 translocation to the cell surface in muscle and adipose cells. [18]. See:
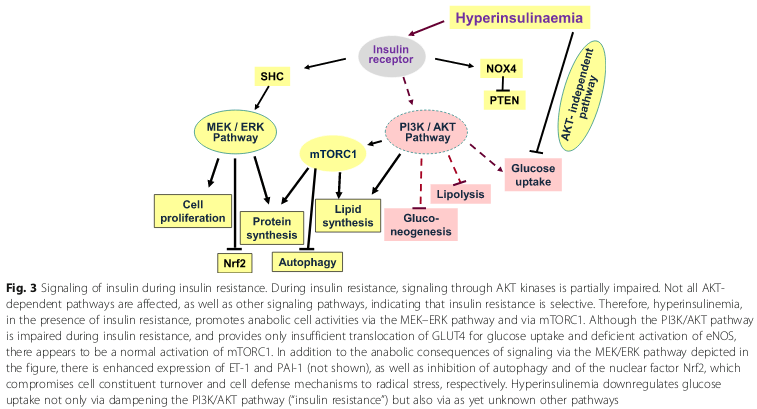
As mentioned, the mechanisms of insulin resistance involve:
(a) impaired glucose transport via post-receptor defects in insulin signaling (i.e., GLUT-4 translocation, and
(b) tyrosine kinase phosphorylation/signaling via the β-subunit of the IR). [14].
Insulin resistance in most cases is believed to be manifest at the cellular level via post-receptor defects in insulin signalling. Despite promising findings in experimental animals with respect to a range of insulin signalling defects, their relevance to human insulin resistance is presently unclear. Possible mechanisms include down-regulation, deficiencies or genetic polymorphisms of tyrosine phosphorylation of the insulin receptor, IRS proteins or PIP-3 kinase, or may involve abnormalities of GLUT-4 function.[15].
Bro-science myths
“Insulin supports exercise-induced fat oxidation by blunting hyperglycemia”
False:
Insulin suppresses fat oxidation [26]; yet, interestingly it is a popular claim, often used to support the practice of slin use during dieting or contest prep, that insulin promotes exercise-induced fat oxidation (by ameliorating hyperglycemia).
There is a weak correlation between insulin sensitivity (a function of glucose and insulin in serum or plasma) & maximal fat oxidation:
Disturbances in fat oxidation have been associated with an increased risk of obesity and metabolic disorders such as insulin resistance. There is large intersubject variability in the capacity to oxidize fat when a person is physically active, although the significance of this for metabolic health is unclear. We investigated whether the maximal capacity to oxidize fat during exercise is related to 24-h fat oxidation and insulin sensitivity. Maximal fat oxidation (MFO; indirect calorimetry during incremental exercise) and insulin sensitivity (Quantitative Insulin Sensitivity Check Index) were measured in 53 young, healthy men (age 24 ± 7 yr, V̇o2max 52 ± 6 ml·kg(-1)·min(-1)). Fat oxidation over 24 h (24-h FO; indirect calorimetry) was assessed in 16 young, healthy men (age 26 ± 8 yr, V̇o2max 52 ± 6 ml·kg(-1)·min(-1)) during a 36-h stay in a whole-room respiration chamber. MFO (g/min) was positively correlated with 24-h FO (g/day) (R = 0.65, P = 0.003; R = 0.46, P = 0.041 when controlled for V̇o2max [l/min]), 24-h percent energy from FO (R = 0.58, P = 0.009), and insulin sensitivity (R = 0.33, P = 0.007). MFO (g/min) was negatively correlated with 24-h fat balance (g/day) (R = -0.51, P = 0.021) but not significantly correlated with 24-h respiratory quotient (R = -0.29, P = 0.142). Although additional investigations are needed, our data showing positive associations between MFO and 24-h FO, and between MFO and insulin sensitivity in healthy young men suggests that a high capacity to oxidize fat while one is physically active could be advantageous for the maintenance of metabolic health. [33].
Recall QUICKI (insulin resistance depends on both blood glucose & insulin concentrations)
Hyperglycemia induced elevated glucose and insulin (insulin resistance) & did not affect fat oxidation rate. The finding that hyperglycemia attenuated fat metabolite levels reflects decreased fat breakdown:
Purpose: The purpose of the present study was to investigate the age-related carbohydrate oxidation and glucose utilisation rate response during exercise at the same relative intensity under hyperglycaemia in aged and young males.
Methods: 16 endurance-trained aged (n = 8; 69.1 ± 5.2 year) and young (n = 8; 22.4 ± 2.9 year) males were studied during 40 min of cycling exercise (60% under both hyperglycemic and euglycemic (control) conditions. Venous blood samples were collected at baseline, post-infusion, mid- and post-exercise. Carbohydrate and fat oxidation rates were determined at both 15 and 35 min during exercise, and glucose utilisation rates were calculated.
Results: The aged group displayed significantly lower rates of carbohydrate oxidation during exercise during maintained hyperglycemia (15 min = 2.3 ± 0.4 vs. 1.6 ± 0.5 g / min; 35 min = 2.3 ± 0.5 vs. 1.5 ± 0.5 g / min) and control (15 min = 2.2 ± 0.4 vs. 1.6 ± 0.7 g / min; 35 min = 1.9 ± 0.7 vs. 1.3 ± 0.7 g / min) conditions (P = 0.01). The rate of glucose utilisation during exercise was also significantly reduced (85.76 ± 23.95 vs. 56.67 ± 15.09 μM / kg / min). There were no differences between age groups for anthropometric measures, fat oxidation, insulin, glucose, NEFA, glycerol and lactate (P > 0.05) although hyperglycemia resulted in elevated glucose and insulin, and attenuated fat metabolite levels.
Conclusion: Our findings highlight that ageing results in a reduction in carbohydrate oxidation and utilisation rates during exercise at the same relative exercise intensity. [34].
Here, the comparison is between aged and young endurance-trained males, and compared hyperglycemia to euglycemia (normoglycemia). In a state of hyperinsulinemia, recall the antilipolytic effects of insulin, which include decreased fat availability and oxidation, and inactivation of hormone-sensitive lipase (HSL), down-regulating mobilization of triglycerides within adipocytes.
“Insulin protects the pancreas by giving the pancreatic islet β-cell a break”
False:
Low-dose insulin glargine (e.g., Lantus) may stave off or slow the progression from prediabetes (impared fasting glucose or impaired glucose tolerance) or early diabetes to type 2 diabetes mellitus (insulin-resistant; T2DM), characterized by total pancreatic islet β-cell failure; but hyperinsulinemia worsens insulin sensitivity. Exogenous insulin in healthy adults worsens insulin sensitivity by increasing HOMA-IR (defined as [fasting values] serum insulin (μU/ml) * plasma glucose (mmol/liter) / 22.5; optimal range 0.5 – 1.4) & worsening QUICKI (defined as 1 / (log(insulin μU/mL) + log(glucose mg/dL))). In healthy adults, Bouche et al. conducted an experiment using an isoglycemic-hyperinsulinemic clamp (mimicking exogenous insulin in healthy adults) and found that pre-exposure to hyperinsulinemia increased insulin secretion by ~40% (21). [27].
In a study authored by Owens, D. R. [27] that is often touted as evidentiary support for the practice of training-day low dose insulin glargine (e.g., Lantus) to promote pancreatic β-cell “rest,” in healthy bodybuilders, the data, rather than supporting this practice, argues for initiating insulin treatment in prediabetes and/or early T2DM before the current observed worldwide clinical practice of initiation of insulin therapy after a duration of T2DM of approximately 10 years x mean HbA1c > 9%. [27].
The study [27] proposed that three (3) groups of individuals are particularly suitable for early intensive insulin therapy:
- Treatment-naive (i.e., no prior exposure to exogenous insulin) patients who present with marked hyperglycemic symptoms with HbA1c > 8.5%
- Latent autoimmune diabetes of adults (type 1.5 diabetics; LADA) patients… characterized by the presence of islet autoantibodies (i.e., treatment initiation indicated when glutamic acid decarboxylase > 20 U/mL) & low C-peptide secretion
- Patients possessing elevated cardiovascular risk factors (e.g., hyperlipidemia, hypertension)…. but the data does not bear out the purported rationale for this practice…
The purported “multifaceted benefits of insulin” that include ostensible anti-inflammatory & antioxidant effects that may protect endothelial function & vascular function is eviscerated by the bodybuilding practice of supra-physiologic anabolic-androgenic steroids that increase ROS (reactive oxygen species) production [31] and deleteriously alter endothelial cell function, particularly those that inhibit 11β-HSD2 (e.g., fluoxymesterone, oxymetholone, testosterone), or alternately inhibit cortisol oxidation and exert MR action (e.g., trenbolone) [30] and combined rhGH use (increasing serum IGF-I) reduces eNOS (endothelial nitric oxide synthase) activity. [32].
These purported benefits are, further, not materialized in any of the large scale studies that measured cardiovascular risk (from ACCORD to ADVANCE to VADT) (29, 30, 31) [27] . In the most relevant and well-powered study, the ORIGIN trial (Outcome Reduction with an Initial Glargine Intervention; a long-term, prospective, large scale, randomized-controlled trial [n=12,537]) in prediabetes and early T2DM patients, patients followed up with at a median of 6.2 years showed no effect on:
- nonfatal myocardial infarction (MI; heart attack)
- nonfatal stroke
- death from cardiovascular causes
- revascularization, or
- hospitalization for heart failure [27]
Indeed, the common bodybuilding use of short & rapid-acting insulin preparations is strongly associated with increased cardiovascular risk.
The crux of the argument that exogenous insulin use promotes “β-cell rest” is rooted in a small (n=7), 2.5 page, 1976 study in diabetic patients that postulated that overcoming glucotoxicity (by exogenous insulin) in turn allows a store of readily available endogenous insulin to be accumulated for early release to a nutrient challenge, thereby, enhancing β-cell function. [29]. See [27] citing Turner, et al. (18). Naturally, the expansion of this logic is inapposite healthy adult bodybuilders using high dose rhGH (i.e., increasing HSL activity, thereby increasing circulating free-fatty acids, inducing insulin resistance).
“Insulin enhances dieting by directing energy to muscle protein synthesis via glycogen synthesis and storage rather than to fat storage in an energy deficit because fat storage is impossible in a deficit”
False:
This claim, often used to promote the practice of slin use during dieting and contest prep, suggests that insulin promotes glycogen storage, and therefore in a state of energy deficit, during its activity peri- workout, slin conserves energy (proceeding from some rudimentary view of conservation of energy), and energy that would otherwise be stored as fat cells is instead dedicated to storage as glycogen and, by this argument’s facile logic, muscle protein.
It cannot be true that insulin use during an energy deficit (dieting) on the one hand (A) cannot synthesize new fat for storage (lipogenesis) & (B) can synthesize new glycogen stores (glycogenesis) and/or new muscle protein (amino acid synthesis/muscle protein synthesis).
Indeed, during caloric restriction, catabolic processes are those pathways directed towards by metabolism; and while slin may not, as an anabolic reaction, induce lipogenesis; it can certainly suppress β-oxidation (fat oxidation) as well as inactivate hormone-sensitive lipase (HSL) activity, down-regulating mobilization of triglycerides within adipocytes) [26] [28], which it does.
References
[1] Thevis, M., Thomas, A., & Schänzer, W. (2009). Insulin. Doping in Sports, 209–226. doi:10.1007/978-3-540-79088-4_10
[2] Biolo, G., Declan Fleming, R. Y., & Wolfe, R. R. (1995). Physiologic hyperinsulinemia stimulates protein synthesis and enhances transport of selected amino acids in human skeletal muscle. Journal of Clinical Investigation, 95(2), 811–819. doi:10.1172/jci117731
[3] Reichlin, S. (1983). Somatostatin. New England Journal of Medicine, 309(25), 1556–1563. doi:10.1056/nejm198312223092506
[4] Sonksen, P. H., Cowan, D., & Holt, R. I. G. (2016). Use and misuse of hormones in sport. The Lancet Diabetes & Endocrinology, 4(11), 882–883. doi:10.1016/s2213-8587(16)30218-2
[5] Rooyackers, O. E., & Nair, K. S. (1997). HORMONAL REGULATION OF HUMAN MUSCLE PROTEIN METABOLISM. Annual Review of Nutrition, 17(1), 457–485. doi:10.1146/annurev.nutr.17.1.457
[6] Biolo, G., & Wolfe, R. R. (1993). Insulin action on protein metabolism. Baillière’s Clinical Endocrinology and Metabolism, 7(4), 989–1005. doi:10.1016/s0950-351x(05)80242-3
[7] Tipton, K. D., & Wolfe, R. R. (2001). Exercise, Protein Metabolism, and Muscle Growth . International Journal of Sport Nutrition and Exercise Metabolism, 11(1), 109–132. doi:10.1123/ijsnem.11.1.109
[8] Wolfe, R. R. (2000). Effects of insulin on muscle tissue. Current Opinion in Clinical Nutrition and Metabolic Care, 3(1), 67–71. doi:10.1097/00075197-200001000-00011
[9] Wolfe, R. R. (2005). Regulation of skeletal muscle protein metabolism in catabolic states. Current Opinion in Clinical Nutrition and Metabolic Care, 8(1), 61–65. doi:10.1097/00075197-200501000-00009
[10] Fujita, S., Rasmussen, B. B., Cadenas, J. G., Grady, J. J., & Volpi, E. (2006). Effect of insulin on human skeletal muscle protein synthesis is modulated by insulin-induced changes in muscle blood flow and amino acid availability. American Journal of Physiology-Endocrinology and Metabolism, 291(4), E745–E754. doi:10.1152/ajpendo.00271.2005
[11] Garlick, P. J., & Grant, I. (1988). Amino acid infusion increases the sensitivity of muscle protein synthesisin vivoto insulin. Effect of branched-chain amino acids. Biochemical Journal, 254(2), 579–584. doi:10.1042/bj2540579
[12] Zhang, X.-J., Chinkes, D. L., Wolf, S. E., & Wolfe, R. R. (1999). Insulin but not growth hormone stimulates protein anabolism in skin wound and muscle. American Journal of Physiology-Endocrinology and Metabolism, 276(4), E712–E720. doi:10.1152/ajpendo.1999.276.4.e712
[13] Fryburg, D. A., Louard, R. J., Gerow, K. E., Gelfand, R. A., & Barrett, E. J. (1992). Growth Hormone Stimulates Skeletal Muscle Protein Synthesis and Antagonizes Insulin’s Antiproteolytic Action in Humans. Diabetes, 41(4), 424–429. doi:10.2337/diab.41.4.424
[14] Cefalu, W. T. (2001). Insulin Resistance: Cellular and Clinical Concepts. Experimental Biology and Medicine, 226(1), 13–26. doi:10.1177/153537020122600103
[15] Wilcox G. Insulin and insulin resistance. Clin Biochem Rev. 2005;26(2):19-39.
[16] Toniolo, L. (2005). Expression of eight distinct MHC isoforms in bovine striated muscles: evidence for MHC-2B presence only in extraocular muscles. Journal of Experimental Biology, 208(22), 4243–4253. doi:10.1242/jeb.01904
[17] Tibaldi, J. M. (2014). Evolution of Insulin: From Human to Analog. The American Journal of Medicine, 127(10), S25–S38. doi:10.1016/j.amjmed.2014.07.005
[18] Kolb H, Kempf K, Röhling M, Martin S. Insulin: too much of a good thing is bad. BMC Med. 2020;1 8(1):224. Published 2020 Aug 21. doi:10.1186/s12916-020-01688-6
[19] Herman, M. E., O’Keefe, J. H., Bell, D. S. H., & Schwartz, S. S. (2017). Insulin Therapy Increases Cardiovascular Risk in Type 2 Diabetes. Progress in Cardiovascular Diseases, 60(3), 422–434. doi:10.1016/j.pcad.2017.09.001
[20] Trommelen, J., Groen, B. B. L., Hamer, H. M., de Groot, L. C. P. G. M., & van Loon, L. J. C. (2015). MECHANISMS IN ENDOCRINOLOGY: Exogenous insulin does not increase muscle protein synthesis rate when administered systemically: a systematic review. European Journal of Endocrinology, 173(1), R25–R34. doi:10.1530/eje-14-0902
[21] Albers, P. H., Pedersen, A. J. T., Birk, J. B., Kristensen, D. E., Vind, B. F., Baba, O., … Wojtaszewski, J. F. P. (2014). Human Muscle Fiber Type–Specific Insulin Signaling: Impact of Obesity and Type 2 Diabetes. Diabetes, 64(2), 485–497. doi:10.2337/db14-0590
[22] Investigators OT (2012). Basal Insulin and Cardiovascular and Other Outcomes in Dysglycemia (ORIGIN Trial). New England Journal of Medicine, 367(4), 319–328. doi:10.1056/nejmoa1203858
[23] Varewijck, A. J., Goudzwaard, J. A., Brugts, M. P., Lamberts, S. W. J., Hofland, L. J., & Janssen, J. A. M. J. L. (2010). Insulin glargine is more potent in activating the human IGF-I receptor than human insulin and insulin detemir. Growth Hormone & IGF Research, 20(6), 427–431. doi:10.1016/j.ghir.2010.10.002
[24] Hadley JS, Hinds CJ. Anabolic strategies in critical illness. Curr Opin Pharmacol. 2002 Dec;2(6):700-7. doi:10.1016/s1471-4892(02)00217-5.
[25] Crown, A. (2000). Characterisation of the IGF system in a primary adult human skeletal muscle cell model, and comparison of the effects of insulin and IGF-I on protein metabolism. Journal of Endocrinology, 167(3), 403–415. doi:10.1677/joe.0.1670403
[26] Newsholme, E., & Dimitriadis, G. (2001). Integration of biochemical and physiologic effects of insulin on glucose metabolism. Experimental and Clinical Endocrinology & Diabetes, 109(Suppl 2), S122–S134. doi:10.1055/s-2001-18575
[27] Owens, D. R. (2013). Clinical Evidence for the Earlier Initiation of Insulin Therapy in Type 2 Diabetes. Diabetes Technology & Therapeutics, 15(9), 776–785. doi:10.1089/dia.2013.0081
[28] Frühbeck, G., Méndez-Giménez, L., Fernández-Formoso, J.-A., Fernández, S., & Rodríguez, A. (2014). Regulation of adipocyte lipolysis. Nutrition Research Reviews, 27(01), 63–93. doi:10.1017/s095442241400002x
[29] Turner, R. C., McCarthy, S. T., Holman, R. R., & Harris, E. (1976). Beta-cell function improved by supplementing basal insulin secretion in mild diabetes. BMJ, 1(6020), 1252–1254. doi:10.1136/bmj.1.6020.1252
[30] Furstenberger, C., Vuorinen, A., Da Cunha, T., Kratschmar, D. V., Saugy, M., Schuster, D., & Odermatt, A. (2012). The Anabolic Androgenic Steroid Fluoxymesterone Inhibits 11 -Hydroxysteroid Dehydrogenase 2-Dependent Glucocorticoid Inactivation. Toxicological Sciences, 126(2), 353–361. doi:10.1093/toxsci/kfs022
[31] Chapa-Dubocq, X., Makarov, V., & Javadov, S. (2018). Simple kinetic model of mitochondrial swelling in cardiac cells. Journal of Cellular Physiology, 233(7), 5310–5321. doi:10.1002/jcp.26335
[32] Ronconi, V., Giacchetti, G., Mariniello, B., Camilletti, A., Mantero, F., Boscaro, M., … Mazzanti, L. (2005). Reduced nitric oxide levels in acromegaly: cardiovascular implications. Blood Pressure, 14(4), 227–232. doi:10.1080/08037050510034293
[33] Robinson SL, Hattersley J, Frost GS, Chambers ES, Wallis GA. Maximal fat oxidation during exercise is positively associated with 24-hour fat oxidation and insulin sensitivity in young, healthy men. J Appl Physiol (1985). 2015 Jun 1;118(11):1415-22. doi: 10.1152/japplphysiol.00058.2015
[34] Malone JJ, Bassami M, Waldron SC, Campbell IT, Hulton A, Doran D, MacLaren DP. Carbohydrate oxidation and glucose utilisation under hyperglycaemia in aged and young males during exercise at the same relative exercise intensity. Eur J Appl Physiol. 2019 Jan;119(1):235-245. doi: 10.1007/s00421-018-4019-4.
About the author
Type-IIx is a physique coach, author, and researcher. Bolus: A Practical Guide and Reference for recombinant Human Growth Hormone Use will be his first published textbook, anticipated for release in early 2023. Ampouletude.com will be Type-IIx's base of operations for coaching services and publications. Type-IIx is proud to be a contributing writer to MesoRx, his home forum, where he is a regular poster.
Leave a Reply
You must be logged in to post a comment.