Estimated reading time: 9 minutes
Table of contents
- Terminology (Key)
- Recomp vs. Partitioning
- Insulin resistance enhances fat loss (decreased FM), but at the expense of retention of LBM (thus is irrational for sane cutting)
- Incretins: GLP-1 and GIP Agonists
- Lipolytic Agents
- Incretins, by Enhancing Insulin Sensitivity, Preserve Muscle Mass (FFMI and Skeletal Muscle Index) while Reducing Fat Mass Index
- Insulin Sensitivity – What Does it Mean? Insulin Resistance ≠ Hyperglycemia
- Exogenous Insulin Causes Insulin Resistance
Recomp (↑LBM and ↓FM) and Incretins (e.g., GLP-1 and GIP agonists like Semaglutide, Tirzepatide) vs. Insulin (exogenous) – Partitioning (the p-ratio) – Leptin and Insulin Sensitivity – Why Hyperglycemia ≠ Insulin Resistance, and How Incretins Preserve Muscle and Preferentially Reduce Fat Stores (i.e., Serving as Partitioning Agents)
Terminology (Key)
- AA: amino acid
- B.F.%.: body fat (as %)
- FA: fatty acid
- FFA: free fatty acid
- FFMI: fat-free mass index (kg/m²; kg = body weight – (B.F. % * body weight) / height squared)
- FM: fat mass
- LBM: lean body mass (FFM: fat free mass, synonym)
Recomp vs. Partitioning
Recomp is a goal or desired outcome of a dietary and training (and drug) regimen, like cutting or bulking. Whereas fat loss (“Cutting”) is defined as decreased FM and retention of LBM, and muscle gain (“Bulking”) is defined as increased LBM and mitigating FM increases, Recomp is defined as increased LBM and decreased FM.
Partitioning is a concept that serves the goal of all rational dietary interventions. The p-ratio (partitioning ratio) describes protein deposited in LBM tissues relative to energy intake and, conversely, protein lost from LBM tissues relative to energy deficit. The p-ratio encompasses the factors of (i) hormone status (i.e., absolute levels of known key hormones), (ii) insulin sensitivity, and (iii) leptin sensitivity. There is an interplay between (i) – (iii).
Insulin sensitivity: when dieting (i.e., in a state of energy deficit), insulin resistance (the reciprocal of insulin sensitivity) enhances fat loss by limiting the muscle’s use of glucose for fuel – sparing glucose for use by the brain and ↑intramuscular FA utilization. When bulking, insulin sensitivity within muscle is good and in fat cells, bad.
Factors that affect insulin sensitivity include [1]:
- Body fat levels; B.F. % (primary predictor): ↑B.F. ⇒ ↑FA substrates for fuel (sparing glucose and protein [that can be used by the liver in gluconeogenesis]) and dictates adipokine signaling (i.e., adipocyte-secreting hormones [Leptin, TNF-α, IL-…, adiponectin, etc.]) ⇒ ↓insulin sensitivity
- Muscular contraction (i.e., activity, as e.g., locomotion, resistance training) ⇒ ↑glucose uptake into muscle cell; GLUT-4 translocation ⇒ ↑insulin sensitivity
- Diet: High carbohydrate (especially refined), saturated fat and low fiber ⇒ ↓insulin sensitivity
- Glycogen repletion or supercompensation ⇒ ↑glucose uptake and glycogenesis ⇒↑insulin sensitivity
- Glycogen depletion (e.g., during the period after an intense training bout, before feeding especially carbohydrates) ⇒ abolition (total depletion) of glucose availability and promotes FA oxidation after exhausting muscle glycogen stores (averaging < 700 g in adults) ⇒ ↑blood (circulating) FFAs ⇒ ↓insulin sensitivity
- Genetic factors partly modifiable by drugs, e.g., in cases of frank hypogonadism, testosterone replacement (TRT) clearly reverses insulin resistance in cases where the etiology of insulin resistance is traceable to testosterone deficiency.
Leptin is an adipokine hormone, secreted primarily by adipocytes (fat cells), that correlates with B.F.%, ↑B.F.% ⇒ ↑Leptin. (visceral vs. subcutaneous depots have different relationships to leptin). At any given B.F.%, women produce ~2 – 3X as much leptin vs. men. Leptin changes with energy restriction and overfeeding. Leptin is a primary energy storage regulatory signal that reflects: (i) B.F. % and (ii) energy intake.
Example 1: Upon initiating a diet, leptin may decline by 50% within 1 week (or less) – although obviously the dieter has not lost 50% B.F. – so acutely, leptin changes become unrelated to B.F. (rather signaling energy intake).
After the initial decline, there is a more gradual decline in leptin related to B.F.% loss.
Example 2: Upon overfeeding, leptin similarly ↑rapidly (i.e., without a relationship to ΔB.F.%, and instead is related to energy intake).
In the short term, leptin secretion is primarily determined by glucose availability – such that pulling glucose out of the fat cell (dieting) ⇒ ↓leptin and vice versa.
Leptin hormone’s site-specific effects include effects on the pancreas and liver, in skeletal muscle it ↑FA and ↓AA and glucose use as fuel substrates (enhancing fat loss, promoting protein sparing)… [1]
Essentially, partitioning (p-ratio) is a concept that couples leptin and insulin sensitivity as the principal factors that determine how changes in caloric intake and macronutrient content affect metabolism (influencing body composition profoundly) as well as hormonal status. We can tweak and enhance it, considering target tissue(s) and our goal (e.g., whether bulking vs. cutting).
Insulin resistance enhances fat loss (decreased FM), but at the expense of retention of LBM (thus is irrational for sane cutting)
First, do not confuse the enhancement of insulin resistance on fat loss with any judgment of insulin resistance being healthy. Insulin resistance, especially in a person who is sedentary, is associated with the metabolic syndrome, diabetes, not to mind central abdominal adiposity, etc.
Insulin resistance is a state wherein the body’s tissues (e.g., liver, pancreas, skeletal muscle) no longer recognizes the body’s insulin and continues to produce glucose in inappropriately high quantities. This state of hyperglycemia is an effect rather than the cause of insulin resistance, though toxic levels of glucose do degrade the pancreatic islet cells’ responsiveness to insulin as one pathway/mechanism of insulin resistance, feeding back to worsen insulin resistance.
Incretins: GLP-1 and GIP Agonists
Fat loss occurs with the GLP-1 and GIP agonists (incretins) — like Semaglutide, and Tirzepatide — that are true insulin sensitizing agents. Still, it is not insulin sensitivity per se that is responsible for fat loss by these incretin drugs – indeed high blood glucose in itself neither enhances fat gain nor inhibits fat loss – but rather, fat loss occurs due to the other effects of these drugs such as altered food preferences, delayed gastric emptying, satiation, that promote appetite control and reduce energy intake.
Incretins (GLP-1 and GIP agonists) directly enhance insulin sensitivity by modulating insulin secretion – coupling it to the presence of elevated glucose concentrations. This insulin secretion subsides as blood glucose concentrations decrease and approach euglycemia. Further, albeit indirectly, by reducing food intake, these drugs result in B.F.% reductions. The reduced B.F. % by reduced food intake then, reduce FM stores (and thereby, circulating FFAs), thereby further enhancing insulin sensitivity.
Lipolytic Agents
In fact, the vast majority of fat loss agents, as lipolytic agents, promote insulin resistance, e.g., β-agonists, stimulant drugs like caffeine and ephedrine, these all either act analogously to or increase the action of catecholamines (epinephrine and norepinephrine, or adrenaline and noradrelanine).
When tissues like the liver and fat cells fail to be stimulated by insulin, glucose is not taken up by the cells. Without glucose, the liver begins to metabolize free-fatty acids (FFAs), thereby increasing ketone levels in the blood, preventing them from being re-esterified into fat cells. In liver and fat cells, without insulin being taken up into these tissues, there is suppression of fat synthesis/lipogenesis (adipocyte) and very-low density lipoprotein (VLDL) synthesis (liver).
Let’s not forget the brain; when systemic blood glucose is elevated, it’ll constantly have its favored energy source and keep us alive. We like that.
Incretins, by Enhancing Insulin Sensitivity, Preserve Muscle Mass (FFMI and Skeletal Muscle Index) while Reducing Fat Mass Index
Making the case for classification of incretins as partitioning agents:
This might all sound perfectly rosy, but we like how the GLP-1 and GIP agonists (e.g., tirzepatide, semaglutide) work because insulin resistance during caloric restriction in skeletal muscle (>60% of body weight, more in bodybuilders) is sort of a scary image, glycogen stores are first catabolized quite rapidly; then intramuscular triglyceride (comprising a mere 1% of wet muscle weight, up to 2% of volume since fat is less dense than skeletal muscle, and ~1/3 of muscle energy since fat is energy dense), and eventually, if the body has to, it will use AA (by catabolizing muscle proteins; proteolysis) for energy to perform work. These agents, then, insofar as they are insulin sensitizing, should serve to promote retention of LBM during cutting.
Indeed, we can see from this Figure from a 2022 research article by Volpe and colleagues [1] that semaglutide quite effectively preserves LBM and preferentially reduces FM with only clinically insignificant reductions in the fat-free mass index (FFMI, kg/m²) and skeletal muscle index during the early adaptation period, that tapers off thereafter:
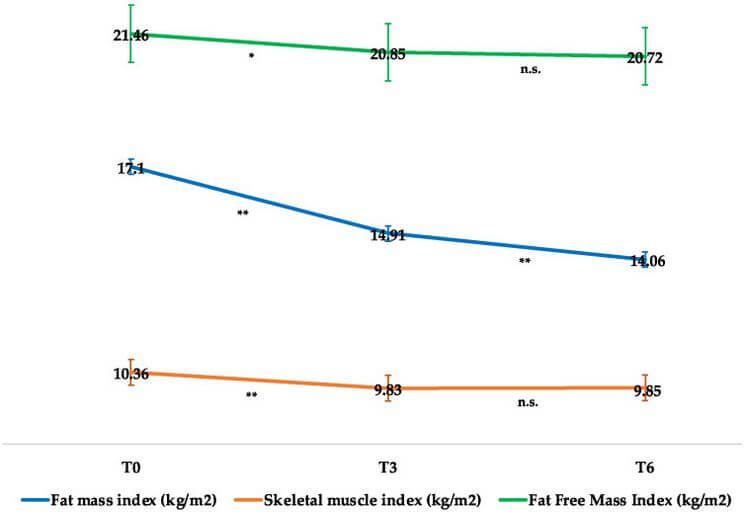
In no small sense, then, by enhancing the insulin sensitivity term of the p-ratio equation, incretins may be fairly classified as partitioning agents, like clenbuterol – but rather than promoting insulin resistance like clenbuterol and its class members, incretins enhance insulin sensitivity and come with far fewer serious side effects.
Insulin Sensitivity – What Does it Mean? Insulin Resistance ≠ Hyperglycemia
It can be very confusing to those familiar with these concepts from bodybuilding discussions that hyperglycemia (elevated blood glucose) is but one factor that is associated with insulin resistance, but is not actually synonymous with insulin resistance (hyperglycemia ≠ insulin resistance). Yes, reducing blood glucose to normal levels is very important in order to improve insulin sensitivity while using exogenous growth hormone (rhGH) because glucose is toxic to the pancreatic β cells. This glucotoxicity at these pancreatic cells results in diminished insulin secretory response to hyperglycemia, thereby fueling the fire of hyperglycemia and glucotoxicity, contributing to insulin resistance – but not constituting the sole etiology of it.
Insulin sensitivityis multifactorial and comprises systemic (e.g., QUICKI) and peripheral (e.g., GLUT-4) components, and is regulated centrally by GLP-1 and GIP. Hyperglycemia is but one factor (the other being insulin) that serves as a proxy for systemic insulin resistance. There are other aspects, including carbohydrate tolerance, etc.
Exogenous Insulin Causes Insulin Resistance
Exogenous insulin reduces blood glucose and thereby prevents this glucotoxicity but actually causes insulin resistance.
Endogenous insulin is secreted in a pulsatile (quick burst) fashion to regulate growth and metabolism, unlike testosterone that is secreted in a more steady-state fashion (gradual release into the blood; but subject to diurnal variations, e.g., more secretion in the morning than midday/evening). Chronic insulin elevations, e.g., those that are germane to the release profile of a daily low dose of insulin glargine (Lantus), possess a relatively large area-under-the-curve (AUC) due to the release profile (high concentrations on long time frames) vs. normal-healthy endogenous insulin release profiles (comparable to regular insulin pharmacokinetics, e.g., Actrapid, Novolin or HumuLin -R). That large AUC of Lantus and/or moderately-high and frequent exogenous regular insulin doses are described as chronic hyperinsulinemia.
This resistance does not occur by negative feedback at the β cell level.
Instead, what occurs with chronic hyperinsulinemia that causes insulin resistance is multifactorial and includes:
- 1. Increasing HOMA-IR and decreasing QUICKI (biochemical measures of insulin resistance and insulin sensitivity, respectively)
- 2. Impaired insulin signal transduction due to receptor (IR) dysfunction and diminished autophosphorylation of the IR, thereby blocking GLUT-4 translocation to the cell surface in muscle and fat cells, meaning more glucose in blood [2]:
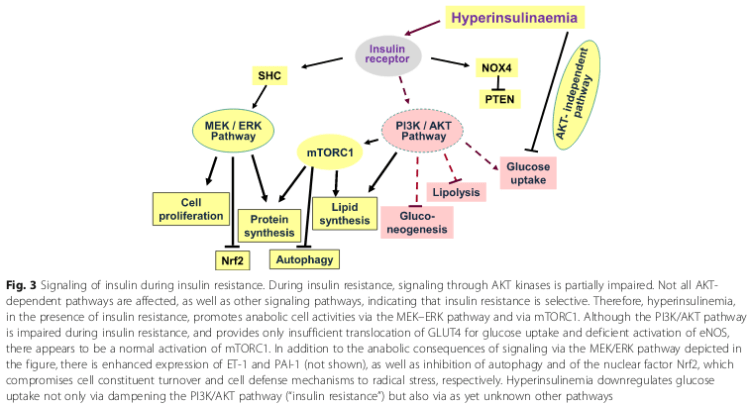
- 3. Increasing sn-1,2-diacylglycerol (DAG) levels and activity due to de novo synthesis.
References
[1] McDonald, L. The Ultimate Diet 2.0: Advanced Cyclical Dieting for Achieving Super Leanness. (2003). Lyle McDonald Publishing.
[2] Volpe S, Lisco G, Racaniello D, Fanelli M, Colaianni V, Vozza A, Triggiani V, Sabbà C, Tortorella C, De Pergola G, Piazzolla G. Once-Weekly Semaglutide Induces an Early Improvement in Body Composition in Patients with Type 2 Diabetes: A 26-Week Prospective Real-Life Study. Nutrients. 2022 Jun 10;14(12):2414. doi: 10.3390/nu14122414.
[3] Kolb H, Kempf K, Röhling M, Martin S. Insulin: too much of a good thing is bad. BMC Med. 2020;18(1):224. Published 2020 Aug 21. doi:10.1186/s12916-020-01688-6
About the author
Type-IIx is a physique coach, author, and researcher. Bolus: A Practical Guide and Reference for recombinant Human Growth Hormone Use will be his first published textbook, anticipated for release in early 2023. Ampouletude.com will be Type-IIx's base of operations for coaching services and publications. Type-IIx is proud to be a contributing writer to MesoRx, his home forum, where he is a regular poster.
Leave a Reply
You must be logged in to post a comment.