
Estimated reading time: 23 minutes
Table of contents
- Introduction
- Purposes
- Thesis
- Illustration of the Perils of Drawing Inferences or Conclusions from Laboratory Bloodwork from Diverse Internet Users; the Divergence in EQ and Primo Estrogenic Effects; and, the Limitations of Circulating Levels as an Index of Tissue-Specific Estrogen Regulation
- Limitations of Circulating Levels as an Index of Tissue-Specific Estrogen Regulation
- Confounding Factors in the Self-Reporting of Estrogenicity from EQ and/or Primo
- Factors that Affect Blood Estrogen Concentrations and Tissue-Level Estrogenic Activities
- Individualized Per-User Factors
- 17β-HSD
- Aromatase
- Symptoms of Low Estrogenicity
- The Cases of EQ and Primo
- Fitting Primo and/or EQ to the Author’s Thesis
- Management of Estrogenicity
- Practical – A Flowchart of Decisionmaking to Address Low Estrogenicity from the use of EQ and/or Primo
- Conclusion
Introduction
Primo and/or EQ Symptoms of Low vs. High Estrogens, Explained, and a Practical Flowchart of Decisionmaking for Symptoms of Low Estrogens as a Result of Primo and/or EQ Use.
So, you have either experience using EQ (boldenone undecylenate; BU), Primo (metenolone enanthate; ME), or perhaps even the two in combination (probably with additional AAS). You have seen reports from users on the internet, or may even have firsthand knowledge of someone for whom the same drugs’ effects were entirely dissimilar from yours. In the first case (internet case reports), you’ve held your tongue – believing that these users most likely administered replacement product (you certainly hope that it wasn’t you!) In the second case, where somebody that you know and understand to have no motivation to deceive you and who is using AAS that is undoubtedly authentic (e.g., authenticated by HP/LC) likewise reports effects that were entirely dissimilar from yours.
WTF is going on here – how can different people experience such markedly different, even opposite, effects from the same drug(s) at similar doses?
Purposes
To –
1. Address this question, in a rigorous way, to reveal to us what was not immediately apparent, and hopefully learn some valuable facts as a result.
2. Provide solutions for those experiencing intolerable symptoms of low estrogenicity as a consequence of the nonmedical use of AAS.
Thesis
The Theory of Compound-Dependent (Per-AAS) and Individualized (Per-User) Estrogenic Potencies (Author’s Model):
Each anabolic-androgenic steroid (AAS)’ effects on estrogenicity (effects associated with ER- α and β activation) depends on Compound-Dependent (Per-AAS) and Individualized (Per-User) Factors that determine both A. actual blood levels of, and B. tissue-level effects of, each AAS’s aromatic products.
The aromatic products produced by AAS run the gamut from nil (i.e., nonaromatizable), to E1 (estrone), a weak estrogen, to E2 (estradiol), a potent (the most potent endogenous) estrogen that virtually all readers will at least know exists and that has an association with classical estrogenic effects (whether E2 is “crashed” or otherwise), to nonendogenous and highly potent estrogens like 7α-methylestradiol (MENT’s remarkably potent aromatic product).
The effects of each AAS (at its dose and duration) and its aromatic products (at their concentrations and durations) dictate the androgen:estrogen (A/E ratio), a proxy for general systemic AAS (direct and side) effects (e.g., gynecomastia). The androgen “arm” of the A/E ratio is a product of the AAS potency to activate AR at its area-under the curve (AUC), as nmol×h/L. The estrogen “arm” of the A/E ratio has two prongs: estrogenic versus antiestrogenic effects. With respect to estrogenic effects, these are a product of the concentration and duration (AUC as nmol×h/L) of the aromatic products (i.e., estrogens) and their potencies to activate ER- α and β. Reciprocally, the antiestrogenic effects that are inherent AAS class effects well established in humans and animals arise out of AAS pituitary (i.e., antigonadotropic) and local tissue (e.g., preventing estrogen uptake) effects, tying back in to the androgen “arm.”
Individualized (per-user) effects of AAS on estrogenicity depend largely on three (3) discrete heritable factors (i.e., a result of one’s genetic phenotype) that are subject to broad inter-individual variation (differences between users) – binding hormone profile¹, 17β-HSD isozyme expression², and Aromatase expression³. Firstly, the user’s binding hormone profile¹ (i.e., SHBG, albumin, α₁ acid glycoprotein, corticosteroid binding globulin activities) determines the activities of free E1/E2 (free estrogens) and free E1/E2:androgen ratio. Secondly, this binding hormone profile¹ interacts with the AAS’s aromatization rate (Vmax) and ester chain length (i.e., logP and hydrophobicity) as the drug concentrations ramp up to a steady-state, influencing the concentration gradient of active estrogens (free E1 and E2) as Esterase frees the parent hormone from the prodrug by active hydrolysis in the whole blood [4]. Thirdly, the user’s 17β-HSD isozyme expression² determines the net flux of E1 (a weak estrogen) vs. E2 (a potent estrogen). Finally, the user’s Aromatase expression³ – partly modifiable by self-regulating body fat mass – determines the absolute concentrations of estrogens (E1 and E2).
Note: Do not be dissuaded just yet by this overwhelming presentation of the Factors that Affect Blood Estrogen Concentrations and Tissue-Level Estrogenic Activities, since we have not yet unpacked these. Continue reading – this author will unpack these factors as we go.
Illustration of the Perils of Drawing Inferences or Conclusions from Laboratory Bloodwork from Diverse Internet Users; the Divergence in EQ and Primo Estrogenic Effects; and, the Limitations of Circulating Levels as an Index of Tissue-Specific Estrogen Regulation
From real internet case reports whose users reported EQ and/or Primo use –
Four (4) distinct cases where no AI was used:
1. Elevations to serum E2 and E1 by 800 mg EQ, 600 mg Tren, 300 mg Test:


EQ (800 mg) + Tren (600 mg) + Test (300 mg) E1 bloodwork Estrone (E1): 1,352 pmol/L (Reference Range: < 250 pmol/L), i.e., 365.6 pg/mL (Very High)
2. Elevations to serum E2 by 300 mg Primo, 300 mg Test:
* Primo + Test low-moderate E2 bloodwork
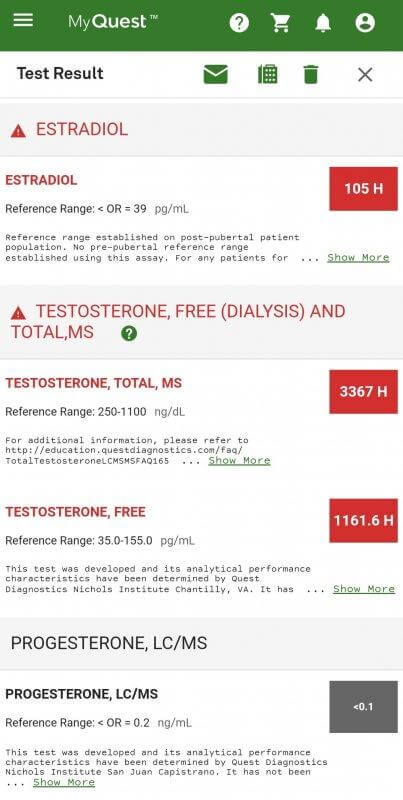
3. Reductions to sub-normal E2 by 750 mg Test, 500 mg EQ, 400 mg Primo:
* Test + EQ + Primo E2 bloodwork
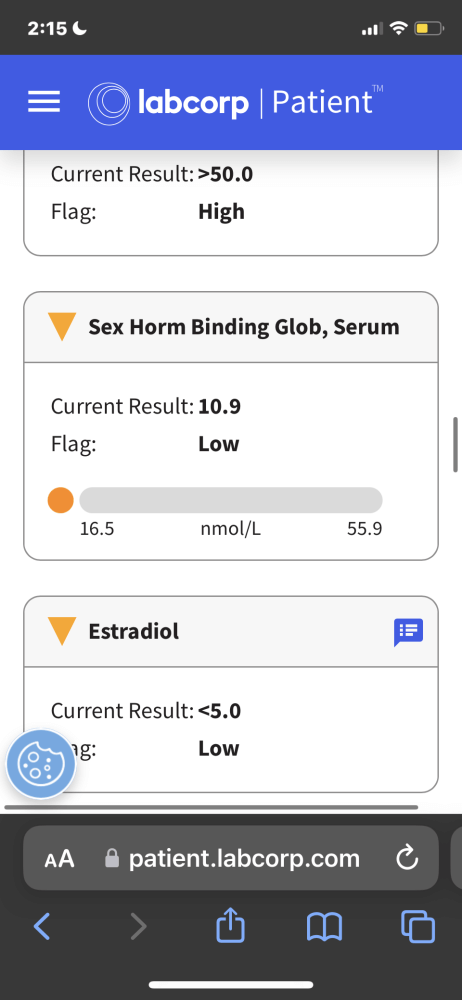
4. Maintained normal E2 by 300 mg Test, 180 mg Primo:
* Primo 180 mg + Test E 300 mg (red) vs. Test C 150 mg (blue)
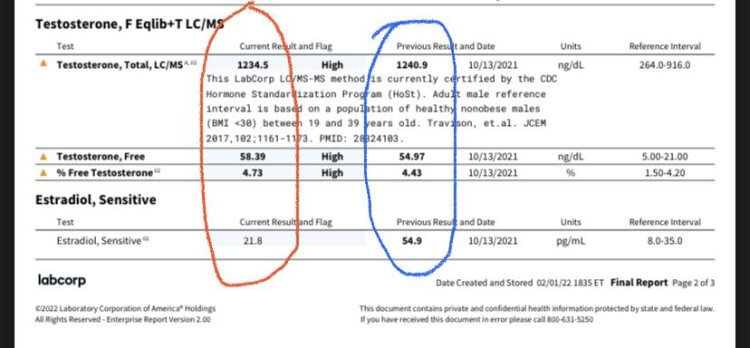
What is the reader to conclude from these data?
That drawing any inferences (not to mind conclusions) from these divergent results is folly. They indicate to us one fact alone – merely, that EQ and/or Primo seem to lower estrogenicity reflected by bloodwork in some instances.
Rather than engender confidence, these results inspire its reciprocal – uncertainty. These bloodwork results illustrate the perils of drawing inferences or conclusions from laboratory bloodwork from diverse internet users.
After the reader understands the Limitations of Circulating Levels as an Index of Tissue-Specific Estrogen Regulation, this author will explain – as parsimoniously as the evidence and the question permits – the Factors that Affect Blood Estrogen Concentrations and Tissue-Level Estrogenic Activities in order to “peek under the hood” at what might be driving this divergence in EQ and Primo estrogenic effects.
Limitations of Circulating Levels as an Index of Tissue-Specific Estrogen Regulation
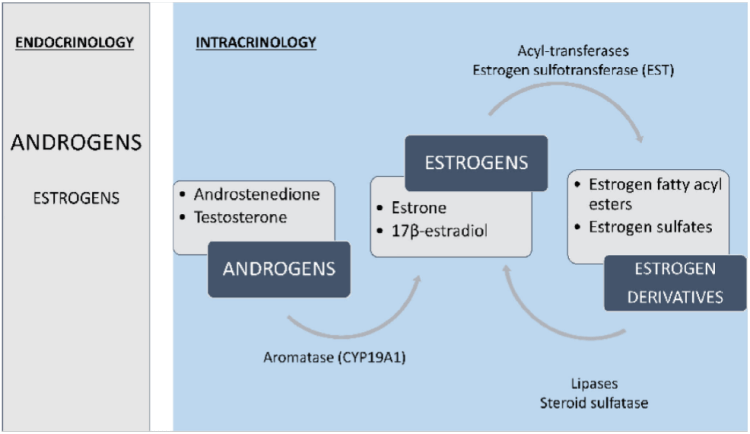
AD: androstenedione
Regulation of estrogen production and metabolism within peripheral tissues is enabled by local expression of Aromatase (CYP19A1), which converts androgens to estrogens (T ⇒ E2 and AD ⇒ E1 [E2 is the most prevalent estrogen in men; which may explain the greater tolerability for EQ in women]). Estrogens further can be converted to estrogen sulfates and estrogen fatty acyl esters via Estrogen Sulfotransferase (EST) and Acyl-Transferases, respectively. Finally, these estrogen derivatives can be converted back to parent estrogens by Steroid Sulfatase (Sulfatase) and Lipase activity [10].
Adipose Tissue (AT) is particularly enriched in estrogen fatty acyl esters and consequently has an extensive buffering system that enables local regulation of estrogen production and metabolism… Notably, in a study of obese men, E2 fatty acyl ester concentrations did correlate in serum and fat (Wang, et al., 2013) [10], possibly indicating that serum estrogen levels influence stored estrogen content in AT, but conversion to bioactive forms is locally regulated [10].
Several clinical studies have demonstrated dissociations between circulating and intra-adipose estrogen levels, including in men (Blankenstein, et al., 1992; Belanger, et al., 2006; Deslypere, et al., 1985; Wang, et al., 2013) [10].
Confounding Factors in the Self-Reporting of Estrogenicity from EQ and/or Primo
This Article will reason through the factors that result in a phenomenon of apparent multiple contradictions – to encompass a reality (i.e., ours) where virtually everyone is telling the truth, administering what they believe are the same drugs at comparable doses, and yet, confoundingly, the estrogenicity (a factor involved in tolerability) differs between individuals. The factors at play are as follows:
1. Laboratory bloodwork analyses may not reflect estrogenicity because tissue-level mechanisms (e.g., blockade of estrogen uptake, intra- vs. endo- crine activities) are involved.
2. Inter-individual variation in binding hormone profile¹, 17β-HSD isozyme expression², and Aromatase expression³ not to mention factors such as ER expression (i..e., density or number) in, e.g., breast tissue (factors that are involved in tolerability).
3. Incompleteness of laboratory bloodwork where EQ is used (i.e., serum E2 measures are insufficient without E1 measures).
4. Counterfeit or replacement product (e.g., Primo is replaced by Test).
5. Differences in ester length (e.g., Bold Cyp vs. EQ) that reflect logP: partition coefficient and lipophilicity: polarity; injection depth (e.g., into the subcutaneous space vs. deep intramuscular) and administration site that differ in bloodflow, and therefore Esterase activity, indirectly influencing the rate of Aromatase reactions.
6. Purported self-reports from fitness professionals that derive an income from generating newsworthy media may be motivated by misrepresentation and/or fraud in order to increase ad revenue at a minimum, if not to integrate their scintillating and novel findings into their portfolio – or “pet concept(s)” – to be used as an insignia or distinguishing mark, upon which their work (e.g., YouTube videos, writings) will be identified and distinguished.
Factors that Affect Blood Estrogen Concentrations and Tissue-Level Estrogenic Activities
Compound-Dependent (Per-AAS) Factors
1. Aromatic products and their potencies to activate ER- α and β (a forthcoming Article will delve into the functions that estrogens serve in men, for the discrete biological functions of these receptors).
a) EQ =[Aromatase]=> E1 (estrone, a weak estrogen, 2% ER-α potency vs E2) and E2 (estradiol, 17β-OH makes it 50-fold more potent than E2) {aromatizes to E1 and E2}.
b) Primo =X[Aromatase] {does not aromatize}, therefore, it does not overcome:
2. Antiestrogenic effects that are class effects of AAS:
a) Inhibition of pituitary-secreted gonadotropins (that indirectly reduce estrogens), and
b) Direct blockade of estrogen activity at the target organ level, preventing estrogen uptake into, e.g., synovial cells, causing “dry, achy joint” symptoms. It is this effect that makes Primo [1], [2] – and before it was discontinued for intolerable androgenic side effects – Masteron [3], so effective for treatment-resistant metastatic breast cancer.
3. EQ’s: a) reduced aromatization rate (Vmax) with respect to boldenone (EQ’s parent hormone).
Km: equal to the concentration of the substrate (abscissa; x-axis values) when the rate is half of the maximum velocity (1/2Vmax; ordinate; y-axis values).
T: testosterone
Aromatization is hindered (vs. T) for androsta-1,4-diene-3-ones (like boldenone; EQ; BU), such that it proceeds slowly [17].
T =[Aromatase]=> E2, Κm = 1.83nM, following Michaelis-Menten kinetics [18].
![T =[Aromatase]=> E2, Κm = 1.83nM, following Michaelis-Menten kinetics](https://thinksteroids.com/wp-content/uploads/2023/07/reaction-rate-vs-substrate-concentration.png)
We do not know the Km for in vivo Aromatase activity with respect to EQ (boldenone undecylenate; BU). However we do know that the Aromatase enzyme is saturable, such that above some dose, that depends on Aromatase expression³ or protein number (and binding hormone profile¹), that more will not cause any further elevations to active estrogens (free E2 and E1). Since EQ (boldenone undecylenate; BU) is subject to hindered aromatization, its reaction rate (Vmax) must be relatively slowed. As such, its in vivo Km must be shifted to the right (vs. that for T/E2) requiring greater concentrations than T for saturation of Aromatase. This tells us that, versus T, higher boldenone doses are needed before Aromatase is saturated (not subject to any increases to E2 at doses above a saturation point).
From [18] we know too that the 40% greater Vmax of Aromatase vis-à-vis T in aged vs. young men was virtually entirely explained by body fat mass and SHBG (i.e., binding hormone profile¹). Since Aromatase is expressed in adipocytes (fat cells) that are subject to increases in number by lipogenesis of new fat cells (adipocytes), maintaining a lifelong low body fat % is an important factor within the reader’s control. Understand that fat cells are not destroyed by caloric restriction – the visual appearance of low body fat after dieting down does not reflect losses of adipocyte number, but merely reduced stores of lipid within those cells. Only lysing or freezing (e.g., chemical lysing like Kybella; CoolSculpting, etc.) for subsequent removal through feces or liposuction (physical removal) of fat cells actually destroy fat cells, such that reduced aromatization would occur.
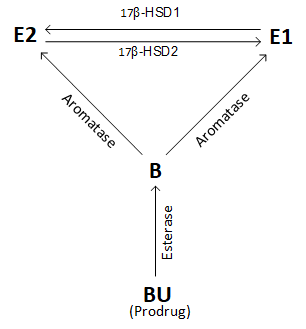
Figure: interconversion of E2 and E1 by 17β-HSD after boldenone undecylenate (BU; EQ) i.m. administration.
Individualized (Per-User) Factors:
4. Depending on an individual’s binding hormone profile¹, the slower release from depot for EQ before reaching steady-state will almost certainly result in reduced Aromatase activity.
5. Depending on an individual’s 17β-HSD isozyme expression², the net flux of estrogens might yield E1 > E2 after EQ, resulting in a far weaker estrogen prevailing in the blood circulation vs. E2.
6. Depending on an individual’s Aromatase expression³, the tolerability of estrogenicity from aromatizable androgen (e.g., EQ) depends in part on the number of Aromatase.
Figure: Molecular target prediction for metenolone (Primo):
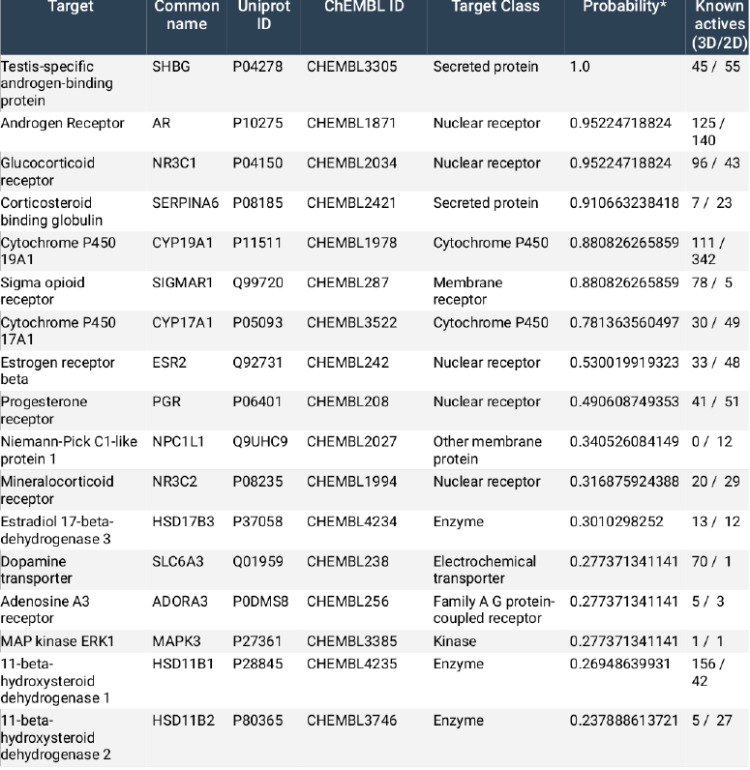
Note: So while there is some evidence (Figure, above) that Primo (metenolone enanthate; ME) has a high probability for Aromatase (Cytochrome P450 19A1) binding (88% probability) – competitive inhibition of which would reduce serum E2 – and low probabilities for binding 17β-HSD1, 17β-HSD2 and 17β-HSD3 – this author will not surmise about these potential mechanisms for effects on estrogenicity, because the model simply does not need them. Besides, we simply do not know what mode of binding it would use nor its biological relevance. It is the exclusive domain of bro-science to engage in such wanton speculation.
Individualized Per-User Factors
Individualized factors (definitions):
- ¹: binding hormone profile: SHBG, albumin, α₁ acid glycoprotein, and corticosteroid binding globulin activities affect inactive bound vs. active free portions of androgens and estrogens.
- ²: 17β-HSD isozyme expression: The relative number of 17β-HSD type 1 vs. type 2 isozymes dictate the relative proportions and absolute levels of circulating E2 and E1, respectively.
- ³: Aromatase expression: The absolute number of Aromatase proteins dictates the levels of aromatic products (i.e., estrogens).
17β-HSD
17β-HSD is a group of enzymes that interconvert steroids (estrogens, androgens) with a keto group in the 17 position (e.g., E1, AD) and those with a hydroxy group in the same position (e.g., E2, T).
All 17β-HSD enzymes catalyze oxidation or reduction of the carbon at position 17 in steroid substrate:
- different substrate substrate preferences (e.g., E1, E2, T, 3β-diol, DHT)
- distinct physiological functions (Jansson, 2009) [15].
Twelve (12) 17β-HSDs have been identified in humans… some catalyze reactions of non-steroidal substrates… if steroid substrate, reaction is either oxidation or reduction, depending on the cofactor and cellular localization [16].
So as to avoid overwhelming the reader with overly complex information, this work will focus on the first two (2) major isoforms of 17β-HSD (type 1, type 2).
17β-HSD1 (type 1), under the control of the A1-Q327 gene, catalyzes reduction steroids (estrogens, androgens) with a 17-keto to one that has a hydroxy group in the same position. So, from E1 (estrone) =[17β-HSD1]=> E2 (estradiol), and from AD =[17β-HSD1]=> T.
17β-HSD1 expression positively correlates to E1 activation and E2 levels [15] and its inhibition reduces. Inhibition 17β-HSD1 => ↓E2 [16].
17β-HSD2 (type 2) reverses the reactions of 17β-HSD1 (i.e., E2 =[17β-HSD2]=> E1 and E3 =[17β-HSD2]=> 16α-hydroxyestrone) and converts T =[17β-HSD2]=> AD (androstenedione), oxidizing the 17-hydroxyl to replace C-17 with a 17-keto group.
Relative over-expression of 17β-HSD2 and under-expression of 17β-HSD1 yields the net effect of increased estrone (E1), subject to inter-individual variation in metabolism.
Aromatase
The Aromatase enzyme, under the control of the CYP19A1 gene, is present in various tissues of humans…including gonads, brain, and adipose tissue (4) [20].
Aromatase is the only human enzyme capable of aromatization of the steroidal A-ring thus converting androgens to estrogens [21].
This enzyme cleaves the 19-methyl from AAS and reconfigures the steroid A-ring such that three, alternating double bonds are formed. This A-ring configuration is described as aromatic (hence, this process is termed aromatization).
In men, there is population variation in height and Aromatase gene expression [22]. This makes sense because estrogens produced by endogenous T’s aromatization to E2 are vital for bone growth and maintenance in men.
Symptoms of Low Estrogenicity
1. “Dry,” aching joints (arthralgia) – Estrogens have naturally anti-nociceptive properties that may be, from a teleological perspective, an evolutionary adaptation to confer pain tolerance during childbirth to women, when estrogen levels are naturally increased [8]. This is believed to be mediated by opioid-containing neurons in the spinal cord that express ER (24) [8]. Animal data shows that ovariectomized mice demonstrate accelerated cartilage turnover (25) that may contribute to reduced joint cushioning [8]. Estrogen suppresses inflammatory cytokine production, and lower estrogen increases levels of inflammatory cytokines such a IL-1 and TNF-α (26)… synovial cells express Aromatase, and when it catalyzes the conversion from androstenedione (AD) to estrone (E1) and estradiol (E2), IL-6 expression is reduced in the joint (28) [8]. Thus, low estrogens, and by corollary AIs, may cause a relative increase in IL-6 production, which is known to act as both a pro- and anti- inflammatory cytokine. It is also known to be one of the key mediators of increased bone loss in post-menopausal women (29) [8].
2. Bone loss – Estrogens play a pivotal role in preventing bone mineral content/density loss. Although androgens have significant effects on male bone, estrogens are more important for bone growth and maintenance… E2 is essential for normal bone mineralization, mass, and turnover; but not for linear bone growth, in men (648, 649) [9].
3. Insulin resistance – Glucose metabolism per kg of muscle is 45% higher in women (756) (likely ER-α-mediated) [9]. In men, testosterone’s beneficial metabolic effects are mediated more by its aromatic product (E2) than by androgen (E2 > T in ↓AT accrual)… ~15% of circulating estrogens derive from testicular synthesis and secretion (Leydig cell) and the remainder by peripheral Aromatase activity… [9].
4. Increased body fat (↑AT; AT: adipose tissue) – In men, E2 regulates body fat stores > T. ERKO: Estrogen Receptor Knockout (ER null) male mice showed AT depots 100% greater by 9 – 12 mo age (aged)… reflects both adipocyte hyperplasia and hypertrophy (281) and accompanied by glucose intolerance and insulin resistance (IR) [9]. ERαKO male mice showed ↑AT inflammation, adipocyte size and impaired glucose tolerance [9].
5. Sexual dysfunction – ER-α signaling in men supports: efferent ductules and epididymal functions; ion transport and H₂O reabsorption necessary to support normal sperm (male reproduction); brain, adipose, skeletal muscle, bone, cardiovascular and immune tissues [9].
Note: While exogenous estrogens cause male reproductive pathologies [9], endogenous estrogens (at normal T levels) are vital for male sexual functioning.
6. ?Reduced skeletal muscle responsiveness to anabolic stimuli – This is not presently supported by the evidence with respect to AAS-induced symptoms of low estrogenicity. Despite being a trope among bodybuilders that AI/SERM use, by antiestrogenic action in skeletal muscle, reduce muscle anabolism; or that very high E2 promotes muscle anabolism – these claims are unsupported by any real evidence (i.e., subjected to rigorous study design and probability and statistical methods to distinguish cause, effect, and random chance). What is supported is that estrogen replacement therapy (HRT, in the literature; different from TRT) increases muscle protein synthesis (MPS) induced by resistance training (RT), but at the expense of basal MPS (i.e., estrogen replacement in post-menopausal women reduces 24-h MPS) [10]… While evidence in ruminants (i.e., cattle) supports exogenous E2 + androgen (e.g., trenbolone acetate), this is, similar to HRT (estrogen replacement) in post-menopausal women, not analogous to AAS in healthy men.
- Since post-menopausal women are aged and typically do not seek HRT (estrogen replacement) for periods of years after cessation of menses, the mere association between low estrogens and attenuated responsiveness to anabolic stimuli is more likely related to other age-related factors than to reduced estrogens (e.g., decreased satellite cell regenerative capacity and diminished IGF-IEc mRNA expression in skeletal muscle).
- Since ruminants do not experience increased IGFBP-1 in response to exogenous E2 like humans [11], which reduces free IGF-I availability and (endogenously) unleashes GH secretion by feedback withdrawal, any estrogen-anabolism connexion in human skeletal muscle is at best tenuous – and probably a mere tertiary factor, related instead to endogenous T and the process of aromatization (augmenting IGF-I) rather than to its aromatic product. Estrogen (e.g., E2) dose-dependently increases IGFBP-1 – which is the reason that women have far greater endogenous GH levels but proportionally lower IGF-I levels than men by body-surface area (a reduced GH response) [13], and why those that are on hormonal contraceptives (i.e., estrogens) must titrate up their rhGH doses in order to see benefits to growth and metabolism, e.g., in adult growth hormone deficiency [14]. In premenopausal women, oral ethinyl estradiol reduces IGF-I levels by up to an average of 30% (24 – 27) [13].
The Cases of EQ and Primo
The use of Primo (metenolone enanthate; ME) and/or EQ (boldenone undecylenate; BU) may result in symptoms of low estrogenicity, that may (or may not) be reflected by sub-normal E2 bloodwork concentrations.
Fitting Primo and/or EQ to the Author’s Thesis
See Theory of Estrogenic Potencies (Author’s Model)):
Each AAS affects the net flux of estrogenicity by its particular effects on blood estrogen concentrations and tissue-level estrogenic activities in the following ways:
Primo:
Primo, as a nonaromatizable AAS, does not produce estrogens. As a result, at moderate/high doses, its effects on the net flux of estrogenicity with respect to aspects of compound-dependent (per-AAS) effects will be markedly anti-estrogenic – the inhibition of pituitary-secreted gonadotropins (that indirectly reduce estrogens in men by suppression of endogenous T synthesis and secretion [steroidogenesis] on which estradiol [E2] biosynthesis depends in men), and direct blockade of estrogen activity at the target organ level, preventing estrogen uptake into cells (e.g., synovial cells, causing “dry, achy joint” symptoms).
EQ:
EQ aromatizes at 58% the rate of testosterone… to estrone (E1) and estradiol (E2) [5]. E1 is a weak estrogen because it lacks the 17β-OH group of E2, possessing a mere 2% of E2’s potency to transactivate ER-α [6]. Since the individual’s 17β-HSD isozyme expression² determines the net flux of E1/E2 balance, it is particularly controlling in the case of EQ effects on net flux of estrogenicity.
EQ is subject to great inter-individual variation vis-à-vis all three enumerated factors (binding hormone profile¹, 17β-HSD isozyme expression², and Aromatase expression³). Its relatively slow Vmax (Aromatase reaction rate), aromatization to E1 (a weak estrogen) and E2, free vs. bound portions thereof, and absolute number of Aromatase are factors in broad divergence of EQ effects on estrogenicity.
Management of Estrogenicity
Visualizing how the user should approach management of estrogenicity should be viewed by a simple model – the inverse U-shaped curve:
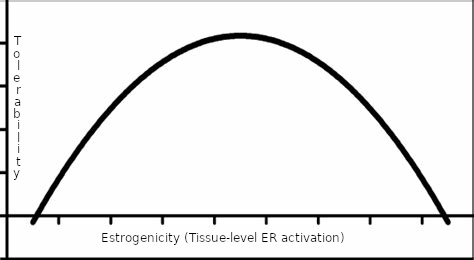
Figure: A simplified model – the U-shaped curve.
The x-axis is related to tissue-level ER activation, that may not be reflected by blood estrogen concentrations. The y-axis reflects tolerability. The area under the curve at its extremes (too low, or too high) is characteristically intolerable. Management of estrogenicity is, then, a “Goldilocks problem.” Estrogenicity cannot be too low or too high, but must be “just right” with respect to tolerability.
The following section is a practical one – a flowchart of decisionmaking for a user to refer to when faced with suspected low estrogenicity (“crashed E2.”)
Practical – A Flowchart of Decisionmaking to Address Low Estrogenicity from the use of EQ and/or Primo
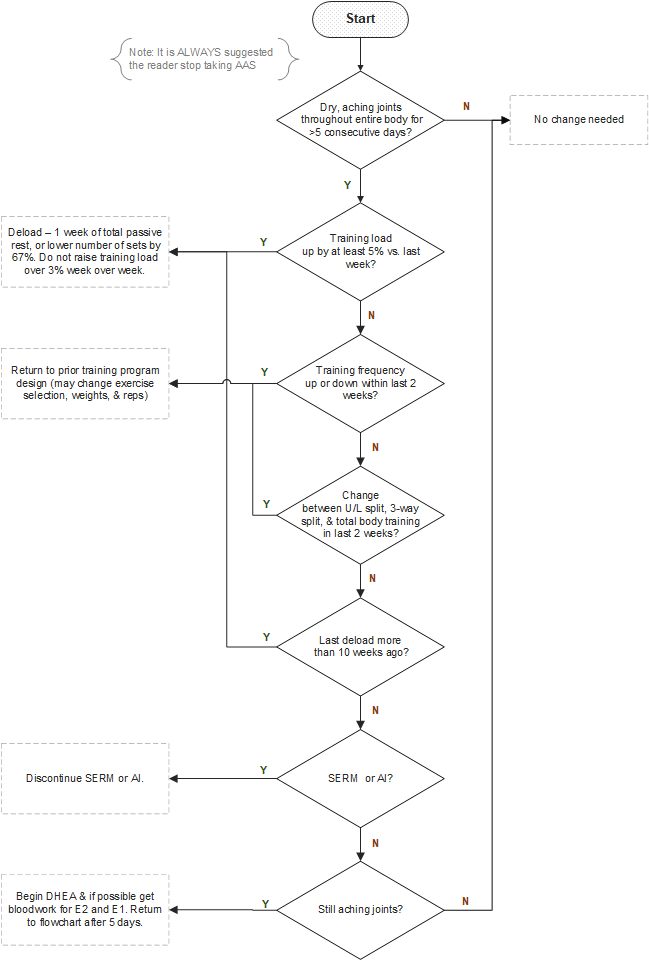
Figure: Flowchart of decisionmaking when faced with Symptoms of Low Estrogenicity.
Conclusion
The estrogenicity (symptoms associated with ER activation) of AAS are subject to per-AAS and per-user effects. Primo, as a nonaromatizable AAS, acts as an antiestrogen and androgen. EQ is an interesting compound for the very fact that it is subject to divergent between-user effects, that depend on factors that include binding hormone profile¹, 17β-HSD isozyme expression², and Aromatase expression³. Laboratory bloodwork analyses miss the mark often for users of AAS seeking to understand estrogenicity due to factors that include local tissue effects and dissociations between intra- and endo- crinology. It is for this reason that self-managing bloodwork and using it to dictate ancillary drug dosing and practices (i.e., polypharmacy) – that are more often bad “bro-science” than medicine – rather than simply staying attuned to the tolerability of these agents, and working through the flowchart diagram presented as needed, leads more often than not to a frustrating game of “whack-a-mole” for AAS users.
References:
[1] Suchowsky, GK, Junkmann, K. [Anabolic steroids and their side-effects]. Acta Endocrinol (Copenh). 1962 Jan;39:68-78. German. PMID: 13918121.
[2] Junkmann, K, Suchowsky, G. [Research on anabolic-active steroids]. Arzneimittelforschung. 1962 Mar;12:214-8. German. PMID: 14452833.
[3] Trams, G. (1977). Effect of drostanolone propionate on the binding of oestradiol and dihydrotestosterone by normal and malignant target tissues. European Journal of Cancer (1965), 13(2), 149–153. doi:10.1016/0014-2964(77)90193-1
[4] Kalicharan, R. W., Bout, M. R., Oussoren, C., and Vromans, H. (2016). Where does hydrolysis of nandrolone decanoate occur in the human body after release from an oil depot? International Journal of Pharmaceutics, 515(1-2), 721–728. doi:10.1016/j.ijpharm.2016.10.068
[5] Gual, C., Morato, T., Hayano, M., Gut, M., and Dorfman, R. I. (1962). Biosynthesis of Estrogens. Endocrinology, 71(6), 920–925. doi:10.1210/endo-71-6-920
[6] Houtman, C. J., Sterk, S. S., van de Heijning, M. P. M., Brouwer, A., Stephany, R. W., van der Burg, B., and Sonneveld, E. (2009). Detection of anabolic androgenic steroid abuse in doping control using mammalian reporter gene bioassays. Analytica Chimica Acta, 637(1-2), 247–258. doi:10.1016/j.aca.2008.09.037
[7] Thomas MP, Potter BV. The structural biology of oestrogen metabolism. J Steroid Biochem Mol Biol. 2013;137:27-49. doi:10.1016/j.jsbmb.2012.12.014
[8] P. Niravath, Aromatase inhibitor-induced arthralgia: a review, Annals of Oncology, Volume 24, Issue 6, 2013, Pages 1443-1449, ISSN 0923-7534, doi.org/10.1093/annonc/mdt037.
[9] Cooke PS, Nanjappa MK, Ko C, Prins GS, Hess RA. Estrogens in Male Physiology. Physiol Rev. 2017 Jul 1;97(3):995-1043. doi:10.1152/physrev.00018.2016.
[10] Rubinow KB. Estrogens and Body Weight Regulation in Men. Adv Exp Med Biol. 2017;1043:285-313. doi:10.1007/978-3-319-70178-3_14
[11] Chidi-Ogbolu N, Baar K. Effect of Estrogen on Musculoskeletal Performance and Injury Risk. Front Physiol. 2019;9:1834. Published 2019 Jan 15. doi:10.3389/fphys.2018.01834
[12] Veldhuis, J. D., and Bowers, C. Y. (2003). Human GH pulsatility: An ensemble property regulated by age and gender. Journal of Endocrinological Investigation, 26(9), 799–813. doi:10.1007/bf03345229
[13] Chanson, P., Arnoux, A., Mavromati, M., Brailly-Tabard, S., Massart, C., … Young, J. (2016). Reference Values for IGF-I Serum Concentrations: Comparison of Six Immunoassays. The Journal of Clinical Endocrinology and Metabolism, 101(9), 3450–3458. doi:10.1210/jc.2016-1257
[14] Cook, D. M., Ludlam, W. H., and Cook, M. B. (1999). Route of Estrogen Administration Helps to Determine Growth Hormone (GH) Replacement Dose in GH-Deficient Adults1. The Journal of Clinical Endocrinology and Metabolism, 84(11), 3956–3960. doi:10.1210/jcem.84.11.6113
[15] He, W., Gauri, M., Li, T., Wang, R., and Lin, S.-X. (2016). Current knowledge of the multifunctional 17β-hydroxysteroid dehydrogenase type 1 (HSD17B1). Gene, 588(1), 54–61. doi:10.1016/j.gene.2016.04.031
[16] Thomas MP, Potter BV. The structural biology of oestrogen metabolism. J Steroid Biochem Mol Biol. 2013;137:27-49. doi:10.1016/j.jsbmb.2012.12.014
[17] Gual, C., Morato, T., Hayano, M., Gut, M., and Dorfman, R. I. (1962). Biosynthesis of Estrogens. Endocrinology, 71(6), 920–925. doi:10.1210/endo-71-6-920
[18] Lakshman, K. M., Kaplan, B., Travison, T. G., Basaria, S., Knapp, P. E., Singh, A. B., … Bhasin, S. (2010). The Effects of Injected Testosterone Dose and Age on the Conversion of Testosterone to Estradiol and Dihydrotestosterone in Young and Older Men. The Journal of Clinical Endocrinology and Metabolism, 95(8), 3955–3964. doi:10.1210/jc.2010-0102
[19] Fouad Mansour M, Pelletier M, Boulet MM, Mayrand D, Brochu G, Lebel S, Poirier D, Fradette J, Cianflone K, Luu-The V, Tchernof A. Oxidative activity of 17β-hydroxysteroid dehydrogenase on testosterone in male abdominal adipose tissues and cellular localization of 17β-HSD type 2. Mol Cell Endocrinol. 2015 Oct 15;414:168-76. doi: 10.1016/j.mce.2015.06.016. Epub 2015 Jun 26.
[20] Attardi BJ, Pham TC, Radler LC, Burgenson J, Hild SA, Reel JR. Dimethandrolone (7alpha,11beta-dimethyl-19-nortestosterone) and 11beta-methyl-19-nortestosterone are not converted to aromatic A-ring products in the presence of recombinant human aromatase. J Steroid Biochem Mol Biol. 2008;110(3-5):214-222. doi:10.1016/j.jsbmb.2007.11.009
[21] Thomas MP, Potter BV. The structural biology of oestrogen metabolism. J Steroid Biochem Mol Biol. 2013;137:27-49. doi:10.1016/j.jsbmb.2012.12.014
[22] Ellis, J. A., Stebbing, M., and Harrap, S. B. (2001). Significant Population Variation in Adult Male Height Associated with the Y Chromosome and the Aromatase Gene. The Journal of Clinical Endocrinology and Metabolism, 86(9), 4147–4150. doi:10.1210/jcem.86.9.7875
About the author
Type-IIx is a physique coach, author, and researcher. Bolus: A Practical Guide and Reference for recombinant Human Growth Hormone Use will be his first published textbook, anticipated for release in early 2023. Ampouletude.com will be Type-IIx's base of operations for coaching services and publications. Type-IIx is proud to be a contributing writer to MesoRx, his home forum, where he is a regular poster.
Leave a Reply
You must be logged in to post a comment.