
Estimated reading time: 14 minutes
Table of contents
Introduction
This is part two of an eleven-part series (2 / 11) that will delve into the unique features of anabolic-androgenic steroids (AAS) by compound or drug. The first part (1 / 11) looked at trenbolone’s unique features.
As with all AAS, stanozolol (“Winny”; Stromba®; StrombaJect®; Winstrol™) has more in common with its counterparts than unique features. But like all of the drugs that this author will review in this series, it has some particular features that “bubble up to the surface” from the ocean of commercially available androgens.
Stanozolol is a member of the heterocyclically-ringed AAS, characterized by a pyrazole ring attachment to its A-ring. Besides extending its duration of biological effects (“half-life”), this chemical modification to the steroidal core exerts other effects that implicate interesting changes unique to this drug:
Stanozolol’s Unique Features
- Mitogenic and myogenic effects by likely increasing free IGF-I bioavailability by decreasing IGFBP-3. [1].
- Glucocorticoid modulation by directly, and indirectly via its 16β-hydroxylated metabolite 16β-ST, negatively regulating the LAGS (low affinity glucocorticoid-binding site) in the liver. [2].
- Antiprogestagenic effects by antagonizing PR (progesterone receptor). [3].
- Profoundly long biological activity (“half-life”) of > 24 h, remarkable among the 17α-alkylated AAS (17AAs). [4].
- Joint aching: Stanozolol, notoriously associated with joint pain (“achy, dry joints”) affects synovial fibroblasts, precursors to cells that comprise the synovial joints (e.g., hip, knees, shoulders), by inhibiting DNA synthesis [5], and perhaps secondarily by (particularly potently) stimulating C1-inhibitor (C1-INH) activity [6] – not unique to stanozolol but common to the 17AAs – that might plausibly decrease vascular permeability and affect joint lubrication and delivery of nutrients to joints. [7].
Mitogenic and Myogenic Effects
Stanozolol decreases IGF-binding protein 3 (IGFBP-3) thereby likely increasing bioavailable IGF-I (free IGF-I)
While stanozolol tends to lower absolute IGF-I, its use in combination with aromatizing androgen and rhGH, that both potently increase it, increases the free, active portion of IGF-I by decreasing IGF binding-protein 3 (IGFBP-3). [1].
Insulin-like growth factor 1 (IGF-I) is a potent growth factor, that is associated with total-body size. IGF-I induces mitosis – cell division – increasing the number of cells in the body’s tissues. IGF-I is, then, mitogenic – it stimulates cell division to “grow everything.” In human skeletal muscle cells, myogenesis is a process whereby new muscle fibers arise out of the fusion of primordial muscle fibers called myoblasts (i.e., skeletal muscle hyperplasia; fusion-dependent mechanisms of human skeletal muscle hypertrophy). [8]. During myogenesis (muscle repair), myoblasts fuse together to form multinucleated myotubes, which eventually mature into fully functioning muscle fibers.
![Figure 1: Satellite cell fusion -dependent vs. -independent mechanisms of human skeletal muscle hypertrophy. [8]. †.](https://thinksteroids.com/wp-content/uploads/2024/07/satellite-cell-fusion-muscle-hypertrophy-750x554.jpg)
The full universe of the commonly used anabolic agents in bodybuilding (e.g., AAS, YK-11 and follistatin [SARMs and/or myostatin inhibitors], clenbuterol [ B₂AR agonists], rhGH, IGF-I and its analogues [peptide growth factors]) additionally induce fusion–independent mechanisms of hypertrophy (muscle remodeling) via the processes of increased protein synthesis and decreased protein catabolism, increasing the cross-sectional area of existing muscle fibers by addition of existing myonuclei.
IGFBPs: A Focus on IGFBP-3 vis-à-vis IGF-I
In humans, there are six (6) IGFBPs that variously modulate IGF-I activity by extending the half-life of the IGF-I and by either potentiating or inhibiting binding of the IGF-I to its receptor (IGF-IR). [9].
In blood circulation, IGF-I is bound to various IGFBPs, particularly the larger IGFBP-3 and its associated acid-labile subunit (ALS), which ↓bioavailability of IGF-I but prolongs its half life in circulation. Binding of IGF-I to ALS prevents transport across the vascular compartment – essentially keeping serum IGF-I elevated because it can’t exit the blood. Conversely, binding of IGF-I to the smaller proteins (IGFBP-1, IGFBP-2, & IGFBP-6) may facilitate penetration into tissues (34). [10].
About 70% of systemic IGF-I circulates in a ~150 kDa ternary (or tertiary) complex, consisting of IGF-I + IGFBP-3 + ALS. [11].
About 20 – 25% of systemic IGF-I is in a smaller (~39 – 44 kDa) binary complex, consisting of IGF-I + IGFBP-3, which can cross the vascular border and may mediate end-target effects. [11].
Less that 10% of systemic IGF-I is in the 7.5 kDa free active form (half-life 12 min). [12].
In human skeletal muscle cells, IGFBP-3 is particularly abundant, and the fibers are saturated further by -BP-2 (regarded as a small inhibitory protein) and lower levels of -BP-4 and -5. [9].
Practical Application
Of the 10 mg average daily production of IGF-I in a healthy normal man (“endogenous levels”), approximately 1 mg (i.e., the free portion in 24-h circulation) exerts the systemic biological effect that we most desire per the free hormone hypothesis. [12].
Unfortunately, the common-sense approach to stimulating the liver’s secretion of IGF-I by administering rhGH presents a conundrum: since GH is the primary stimulator of IGFBP-3 and ALS [11], exogenous supraphysiologic GH then increases the total fraction of free IGF-I while reducing its relative bioactivity, analogously to free testosterone vis-à-vis supraphysiologic testosterone (via increased albumin), ergo free androgen and AAS.
It is this author’s thesis, then, that stanozolol may be fairly viewed as an agent to increase IGF-I bioavailability vis-à-vis rhGH a la mesterolone (Proviron®) vis-à-vis AAS. But while the latter’s effect on SHBG is fairly impotent versus the per se androgenic effect on it (i.e., to potently reduce it), stanozolol should fare better by comparison for practical application.
Glucocorticoid Modulation
Stanozolol negatively regulates the low affinity glucocorticoid-binding site (LAGS)
In the liver, the LAGS is a low affinity binding site for glucocorticoids like cortisol that loosely binds free circulating catabolic hormones. Only stanozolol has been demonstrated to provoke a time- & dose- dependent inactivation of the LAGS-binding site that was irreversible, that suggests negative allosteric modulation of glucocorticoid binding to the LAGS. [13].
Stanozolol most potently among the 17AAs (stanozolol > fluoxymesterone [“Halo”; Androxy®; Ora-Testryl®; Ultandren®] > metandienone [“Dianabol”] > methyltestosterone):
- ↓affinity of glucocorticoids (e.g., cortisol)
- ↓ # of LAGS-binding sites
- ↑ dissociation rate of glucocorticoids from the glucocorticoid binding sites. [13].
Stanozolol’s novel mechanism versus the other 17AAs that makes it not merely particularly potent but unique in its irreversible inactivation of the LAGS-binding site is that this action is mediated by its 16β-hydroxylated metabolite (16β-ST). [14].
Practical Application
But here’s the “rub” – the net systemic consequence of this negative allosteric regulation by stanozolol is an effective increase in classical GR-signaling (i.e., increased skeletal muscle catabolism) by increasing glucocorticoid availability to the cytosolic GR. [14].
This unique feature of stanozolol is, then, unfavorable. ✖ enhanced muscle catabolism (Not a Good Thing™).
Antiprogestagenic Effects
Stanozolol antagonizes the progesterone receptor (PR)
Stanozolol is a remarkably potent PR inhibitor, exhibiting ½ its efficacy at a low 0.1188 uM concentration in the following assay of small molecule progesterone inhibitors. [3].:
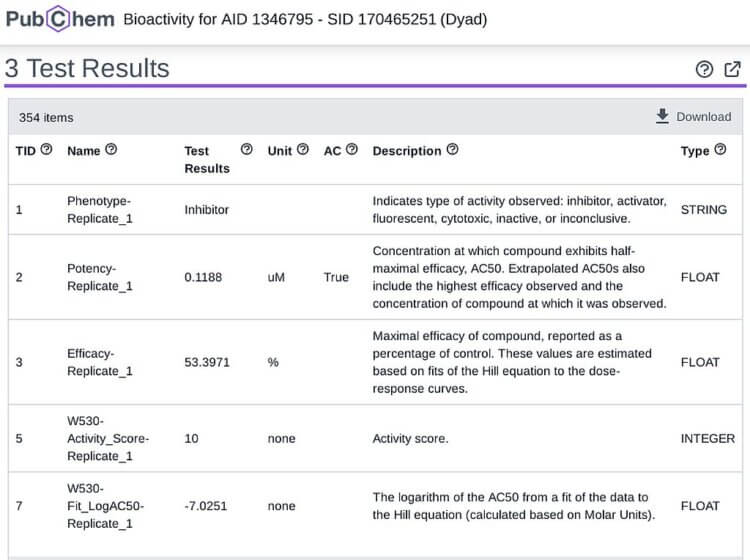
Practical Application
In July of 2022, I published a quasi-article in a post on the community form of MESO-Rx titled Article on distinguishing progestins, prolactin, and progestagenic androgens (e.g., Tren, MENT, Deca) & SERM vs. AI logic [by Type-IIx] to disambiguate confusion between progestins, prolactin, and progestagenic androgens. In it, I summarize that progestagenic androgens (e.g., MENT [trestolone], oxymetholone [“Adrol”; “Drol”]) tend to aggravate gynecomastia and contribute to more potent HPG axis suppression (“shutdown” of endogenous testosterone secretion) by altering KNDy dendron pulsatility, can exert tissue-level antiandrogenic (inhibition of AR) effects, and can contribute directly to gynecomastia (as well, estrogens upregulate PR synthesis) [see article link for citations].
Rather than relying solely on an aromatase inhibitor (AI) drug to reduce absolute estrogen levels to combat these maladies (i.e., “shutdown” and “gyno”), a rational drug selection might include stanozolol.
Toxicity, but also, Duration of Biological Effects
Stanozolol has a remarkably long > 24 hour biological half-life
Definitions and Concepts
- Biological half-life: A single-dose pharmacokinetic concept, a measure of time (h, min, sec) that describes the duration that a drug remains active in the body after ingestion (i.e., p.o., swallowing) or parenteral (e.g., i.m., i.v.) administration. It is measured as the time at which a drug’s concentration is reduced to ½ its original concentration and reflects metabolism and/or excretion.
- Terminal half-life: A multi-dose pharmacokinetic concept, a measure of time (h, min, sec) that describes the duration that a drug remains active in the body after all dosing has stopped (i.e., after a blast or cycle). It is a measurement of how long a drug remains, even if inactive, in the body after all dosing has stopped and in our case (the case of injecting esterified AAS designed to increase blood levels to equilibrium over terms of weeks) is far longer than the biological half-life of the drug.
Contextualizing the Data
The 17AA drugs (17α-alkylated; e.g., stanozolol, metandienone, oxymetholone) are characterized by relatively long biological half-lives (in the order of hours) due to the addition of the methyl group in the α-oriented plane of the 17th carbon of the classical steroidal core or sterane ring. This 17α-alkylation contributes to prolongation of the anabolic effect via hepatic metabolism, and inhibits aromatization of the A-ring to estrogens.
Like this modification at C-17, the modification made to stanozolol – a pyrazole ring addition, fused to the juncture between the second and third carbons of ring-A of the 5α-dihydro sterane ring – serves to prolong the anabolic effect via hepatic metabolism even further still. Whereas methyltestosterone’s half-life ranges from approx. 3 to 7 h depending on the source of authority (i.e., Hazardous Substances Data Bank [HSDB] vs. DrugBank), fluoxymesterone’s oddly precise 9.2 h biological half-life explains its common prescription guidelines (Androxy® pamphlet) for adults to be used at a dose of 10 through 40 mg daily in 1 through 4 doses, i.e., every 4 hours), and even taking the inapplicable intravenous (i.v.) data for dehydrochloromethyltestosterone (Oral-Turinabol®; “OT”; “Tbol”) showing its 16 h terminal half-life (parenteral administration prolongs bioactivity), stanozolol’s > 24 h biological half-life is striking since it’s 50% > that of OT’s terminal half-life after intravenous infusion.
The 17AAs’ hours-long biological half-lives are considerably longer than ingested (p.o.; swallowed) base hormone. For example, guzzling a vial of testosterone intended for injection isn’t going to give you anything in terms of gains because of its rapid metabolism and excretion; nor will it be particularly toxic (though I do not recommend it). A medical hypothesis published in the eponymous journal by doctors Peter Bond, William Llewellyn, and Peter Van Mol presents a compelling case for 17AA hepatotoxicity as a function of AR activation of increased reactive-oxygen species (ROS) via mitochondrial fatty-acid β-oxidation in liver cells, i.e., hepatotoxicity (AU) ≈ AR transactivation potency (RLU) × t½/(ke * Vd) × dose (mg), or more basically stated – a drug’s hepatotoxicity is the product of its anabolic potency and resistance to hepatic breakdown.
Practical Application
Stanozolol is therefore a particularly hepatotoxic drug despite its clinical use! Practically, however, one need not fret about dose optimization – taking it once daily while on a regimen guarantees round-the-clock anabolism. Its hepatotoxicity suggests that drinking alcohol to excess while using it is unwise and that a blast or cycle of it might be best limited to terms of weeks.
Joint Pain
Stanozolol and Joint Pain, Redux
This author has previously discussed stanozolol’s proclivity to cause joint pain in the Meso-Rx Article titled, “Anabolic Steroids and Growth Hormone: Their Impact on Bones, Tendons, Ligaments, and Joints”. This article’s subsections will succinctly describe these topics.
Synovial cell DNA synthesis inhibition
Stanozolol inhibits DNA synthesis in the synovial cells that comprise the cartilage of major joints like the hips, knees, and shoulders. [5]. Inhibiting DNA synthesis results in slowed tissue regeneration rate and capacity. Since these tissues are already characterized by relatively slow rates of repair – hence the need for surgical intervention when catastrophically injured – versus skeletal muscle, this results in more fragile, less robust sinew in the major joints while using stanozolol.
Increased C1-INH activity decreases vascular permeability that may reduce joint lubrication and delivery of nutrients to joints
Stanozolol’s potency to increase C1 inhibitor (C1-INH) activity is the reason for its efficacy to treat hereditary angioedema (HAE), a condition marked by mucosal swelling that leaves its patients suffering with tissues bloated by fluids. [7]. C1-INH is a multi-serine protease inhibitor that controls several catalytic pathways including classical component activation. [7]. HAE results from ↓C1-INH. C1-INH suppresses bradykinin, that via its action at B₂R mediates vasodilation and enhances permeability resulting in angioedema. [7]. When C1-INH is decreased, ↑vascular permeability ensues (subcutaneous & submucosal) – causing angioedema – due to ↑bradykinin (which C1-INH attenuates) [due to effects on classical contact system & complement activation]. [7]. Stanozolol up-regulates C1-INH gene expression & plasma aminopeptidase P (catabolizing kinins) (55, 56). [7]. Stanozolol probably enhances hepatic C1-INH production by direct hepatic action rather than AR action per se. [16].
For a normal (i.e., healthy) adult user of stanozolol, this increased C1-INH likely decreases C1-INH to subnormal levels, thereby decreasing vascular permeability, that in turn reduces the delivery of fluid and nutrients to the joint required for healthy functioning.
Practical Application
The wrapping of joints using when performing heavy lifting during resistance training bouts or sessions is wise during a stanozolol course. Since aromatase inhibitor (AI) drugs tend to induce arthalgia (joint pain) by increasing inflammatory cytokine production in the synovial cells of these joints (28) perhaps modulating AI dose and compound selection to symptomology and stanozolol tolerability is wise. [17].
Conclusion
Stanozolol is a classic “hardening” agent and attenuated androgen possessing reduced androgenic features and is widely regarded as appropriate for female use among the universe of AAS. Its combination with rhGH is rational for its increasing bioactive IGF-I, the free portion, by reducing IGFBP-3. Its unfavorable effects on glucocorticoids suggests its rational combination with drugs that enhance glucocorticoid activity for muscle anabolism (e.g., trenbolone). Its antigestagenic effects suggests its rational combination with drugs that are gestagenic (e.g., oxymetholone). Its toxicity calls for abstaining from intoxicating liquor in excess but by that same feature (resistance to hepatic breakdown), a reduced dosing frequency as well. Finally, its effects on joints calls for wrapping them during heavy lifting and modulating AI use to tolerability and symptomology.
Stanozolol is quite a unique AAS! Of course, like any intervention such as drug use, there are inherent tradeoffs – positives and negatives to be balanced to one’s objective, be it to recomp, cut, or bulk.
Footnotes
†: Figure 1 – , herein, depicts a dismally incomplete (one might say thoroughly pigeonholed) understanding of anabolic agents – with the effect of diminishing their illustrated potency – by the authors responsible for this illustration. [8]. Rather than dichotomous, there is substantial overlap (a multi-, at least two- pronged flow from AAS, MSTN deficiency, clenbuterol and Akt overexpression) between fusion- independent and dependent mechanisms, contrary to this depiction. Moreover, it fails to include the IGFs and GH in fusion-dependent contributors. This author refers the reader to Figure 2 from Dubois, V., Laurent, M., Boonen, S., Vanderschueren, D., & Claessens, F. (2011). Androgens and skeletal muscle: cellular and molecular action mechanisms underlying the anabolic actions. Cellular and Molecular Life Sciences, 69(10), 1651–1667. doi:10.1007/s00018-011-0883-3. MyomiRs (e.g., miR- 133, 206, 221, 222) are not the sole avenue of overload-induced hypertrophy anyhow, and certainly do not run totally parallel to, as if sealed-away from, AAS mechanisms! That fact should “jump out” to the most astute reader.
References
[1] Fryburg, D. A., Weltman, A., Jahn, L. A., Weltman, J.Y., Samojlik, E., Hintz, R. L., & Veldhuis, J. D. (1997). Short-Term Modulation of the Androgen Milieu Alters Pulsatile, But Not Exercise- or Growth Hormone (GH)-Releasing Hormone-Stimulated GH Secretion in Healthy Men: Impact of Gonadal Steroid and GH Secretory Changes on Metabolic Outcomes1. The Journal of Clinical Endocrinology & Metabolism, 82(11), 3710–3719. do i:10.1210/jcem.82.11.4379
[2] Betancor-Hernández, E., Pérez-Machı́n, R., Henrı́quez-Hernández, L., Mateos-Dı́az, C., Novoa-Mogollón, J., & Fernández-Pérez, L. (2003). Photoaffinity labeling identification of thyroid hormone-regulated glucocorticoi d-binding peptides in rat liver endoplasmic reticulum: an oligomeric protein with high affinity for 16β-hydroxylated stanozolol. The Journal of Steroid Biochemistry and Molecular Biology, 87(4-5), 253–264. doi:10.1016/j.jsbmb.2003.09.009
[3] National Center for Biotechnology Information. “PubChem Bioassay Record for Bioactivity AID 1346795 – SID 170465251, Source: Tox21” PubChem, pubchem.ncbi.nlm.nih.gov/bioassay/1346795#sid=170465251. Accessed 23 June, 2024.
[4] DrugBank 6.0: the DrugBank Knowledgebase for 2024. “Stanozolol, Record: DB06718” DrugBank, drugbank.ca/drugs/DB06718. Accessed 23 June, 2024.
[5] Ellis AJ, Cawston TE, Mackie EJ. The differential effects of stanozolol on human skin and synovial fibroblasts in vitro: DNA synthesis and receptor binding. Agents Actions. 1994 Mar;41(1-2):37-43. doi: 10.1007/BF01986391
[6] Sloane, D. E., Lee, C. W., & Sheffer, A. L. (2007). Hereditary angioedema: Safety of long-term stanozolol therapy. Journal of Allergy and Clinical Immunology, 120(3), 654–658. doi:10.1016/j.jaci.2007.06.037
[7] Cugno, M., Zanichelli, A., Foieni, F., Caccia, S., & Cicardi, M. (2009). C1-inhibitor deficiency and angioedema: molecular mechanisms and clinical progress. Trends in Molecular Medicine, 15(2), 69–78. doi:10.1016/j.molmed.2008.12.001
[8] Solsona R, Pavlin L, Bernardi H, Sanchez AM. Molecular Regulation of Skeletal Muscle Growth and Organelle Biosynthesis: Practical Recommendations for Exercise Training. (2021). Int J Mol Sci. 22(5):2741. doi: 10.3390/ijms22052741
[9] Foulstone, E. J., Savage, P. B., Crown, A. L., Holly, J. M. P., & Stewart, C. E. H. (2003). Role of insulin-like growth factor binding protein-3 (IGFBP-3) in the differentiation of primary human adult skeletal myoblasts. Journal of Cellular Physiology, 195(1), 70–79. doi:10.1002/jcp.10227
[10] Hadley JS, Hinds CJ. Anabolic strategies in critical illness. Curr Opin Pharmacol. 2002 Dec;2(6):700-7. doi:10.1016/s1471-4892(02)00217-5
[11] Kelley, K. M., Oh, Y., Gargosky, S. E., Gucev, Z., Matsumoto, T., Hwa, V., … Rosenfeld, R. G. (1996). Insulin-like growth factor-binding proteins (IGFBPs) and their regulatory dynamics. The International Journal of Biochemistry & Cell Biology, 28(6), 619–637. doi:10.1016/1357-2725(96)00005-2
[12] Guler, H.-P., Zapf, J., Schmid, C., & Froesch, E. R. (1989). Insulin-like growth factors I and II in healthy man. European Journal of Endocrinology, 121(6), 753–758. doi:10.1530/acta.0.1210753
[13] Fernández, L., Chirino, R., Boada, L. D., Navarro, D., Cabrera, N., del Rio, I., & Díaz-Chico, B. N. (1994). Stanozolol and danazol, unlike natural androgens, interact with the low affinity glucocorticoid-binding sites from male rat liver microsomes. Endocrinology, 134(3), 1401–1408. doi:10.1210/endo.134.3.8119180
[14] Betancor-Hernández, E., Pérez-Machı́n, R., Henrı́quez-Hernández, L., Mateos-Dı́az, C., Novoa-Mogollón, J., & Fernández-Pérez, L. (2003). Photoaffinity labeling identification of thyroid hormone-regulated glucocorticoid-binding peptides in rat liver endoplasmic reticulum: an oligomeric protein with high affinity for 16β-hydroxylated stanozolol. The Journal of Steroid Biochemistry and Molecular Biology, 87(4-5), 253–264. doi:10.1016/j.jsbmb.2003.09.009
[15] Bond P, Llewellyn W, Van Mol P. Anabolic androgenic steroid-induced hepatotoxicity. Med Hypotheses. 2016 Aug;93:150-3. doi: 10.1016/j.mehy.2016.06.004
[16] Sloane, D. E., Lee, C. W., & Sheffer, A. L. (2007). Hereditary angioedema: Safety of long-term stanozolol therapy. Journal of Allergy and Clinical Immunology, 120(3), 654–658. doi:10.1016/j.jaci.2007.06.037
[17] P. Niravath, Aromatase inhibitor-induced arthralgia: a review, Annals of Oncology, Volume 24, Issue 6, 2013, Pages 1443-1449, ISSN 0923-7534, doi.org/10.1093/annonc/mdt037.
About the author
Type-IIx is a physique coach, author, and researcher. Bolus: A Practical Guide and Reference for recombinant Human Growth Hormone Use will be his first published textbook, anticipated for release in early 2023. Ampouletude.com will be Type-IIx's base of operations for coaching services and publications. Type-IIx is proud to be a contributing writer to MesoRx, his home forum, where he is a regular poster.
Leave a Reply
You must be logged in to post a comment.