OhNoYo
New Member
When Will We Have a SAFE & EFFECTIVE Myostatin Inhibitor in the Future?
Anyone???
 opcorn:
opcorn:
Anyone???
opcorn:
Last edited by a moderator:
MESO-Rx
Anabolic Steroids
Follow along with the video below to see how to install our site as a web app on your home screen.
Note: This feature may not be available in some browsers.
opcorn:")
if they ever make one that is real and works for real - steriods will no longer have a place in bodybuilding guys!
it is the truth - why mess with ur hormones when you could just alter ur body to create more muscle cells - haha but keep wishing nothing is real so far good at all that i know of - there is that dog and a couple animals and people with the gene issue
- there is 1 german boy that has double muscle and has the myostin gene problem - i wish i could find him online but i can find no new pix other than him as a baby - he COULD BE THE BEST BOSYBUILDER EVER NO JOKE THINK ABOUT IT


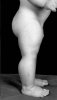
I was surprised to not see more posts on this thread. After all, this is an area of active R&D and undoubtedly leads to increased musculature. Is it possible that many do not recall one the images first published (below)? This image is from the 1997 PNAS article, "McPherron AC, Lee S-J. Double muscling in cattle due to mutations in the myostatin gene. Proceedings of the National Academy of Sciences of the United States of America 1997;94(23):12457-61." http://www.pnas.org/content/94/23/12457.full.pdf
The original publication was in the same year that included the above authors, "McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 1997;387(6628):83-90." Regulation of skeletal muscle mass in mice by a new TGF-p superfamily member In this publication, we were introduced to the "Mighty Mouse."
Myostatin, a member of the transforming growth factor (TGF)-? superfamily, plays a potent inhibitory role in regulating skeletal muscle mass. Increasing size and strength of skeletal muscle represents a promising therapeutic strategy for muscular disorders. Inhibition of myostatin by gene disruption, transgenic expression of myostatin propeptide, or injection of propeptide or myostatin antibodies, causes a widespread increase in skeletal muscle mass. Several peptides, in addition to myostatin propeptide and myostatin antibodies, can bind directly to and neutralize the activity of myostatin. [More on this later.]

i disagree. To most, bodybuilding is about pushing the abnormal envelope. So if steroids are good...and an effective (and attainable) myostatin inhibitor is good...then 1+1=3, right? 400lb behemoth bodybuilders here we come!
Doesn't flex wheeler claim to have a myostatin defect? :d
Here is the dog
so you can see steriods would not matter much at all - this dog does not take riod or even bodybuild and is pure muscle - so imagine if someone just ate clean and bodybuilded clean with this condition - they would be 300 lbs more ripped than jay cutler for real \
and here is the german child - he has muscle legs at 7 moths guys - think if he bodybuilds at 20 years old!!!!!
How myostatin inhibition could be incorporated into the doping toolkit is unknown, but there are concerns that genetic manipulation of myostatin gene expression may be in the first wave of ‘‘gene doping.’’ Currently, the potential effects of anti-myostatin treatments on athletic performance are unclear, but a growing body of data has shown that commensurate increases in muscle strength and power may not accompany the cellular and histological changes resulting from inhibited myostatin function. This review highlights the function of myostatin in muscle growth, developments in myostatin-targeted pharmacological and gene therapies, and the potential of these therapies to be appropriated by unscrupulous athletes and their entourages looking for a short cut to excellence.
Fedoruk MN, Rupert JL. Myostatin inhibition: a potential performance enhancement strategy? Scandinavian Journal of Medicine & Science in Sports 2008;18(2):123-31. Myostatin inhibition: a potential performance enha... [Scand J Med Sci Sports. 2008] - PubMed result
A decade has passed since myostatin was first identified as a negative regulator of muscle growth. Since then, studies in both humans and animals have demonstrated that decreasing the levels of this growth factor or inhibiting its function can dramatically increase muscle size, and a number of therapeutic applications of myostatin inhibition to the treatment of myopathies and muscle atrophy have been proposed. As such treatments would be likely to also stimulate muscle growth in healthy individuals, there is a growing concern among anti-doping authorities that myostatin inhibitors may be among the next generation of ergogenic pharmaceuticals or even in the vanguard of “gene doping” technology. While the ability to stimulate muscle growth through myostatin inhibition is well documented, a growing body of evidence suggests such increases may not translate into an improvement in athletic performance. This article briefly reviews the function of this potent regulator of muscle development and explores the potential therapeutic uses, and potential ergogenic abuses, of myostatin manipulation.

 )]opcorn:
)]opcorn:Doesn't the peptide follistatin ( sp?) have an effect on myostatin ?
Have a buddy who has gone through a few cycles of it, noted soreness in the injection spot and improved recovery. I'm too chicken to try something that new ( and I have cancer in my family)