Hypogonadism with Two Novel Mutations of the Luteinizing Hormone ?-Subunit Gene Expressed in a Compound Heterozygous Form
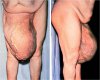
A 31-yr-old, 46,XY man was referred for investigation of sexual infantilism. He was 182 cm tall, weighed 89 kg, had an arm span of 187 cm, and his waist circumference was 108 cm. He was phenotypically male at birth, with descended testes. He had bilateral gynecomastia, scant normally distributed pubic and axillary hair (Tanner stage 2), and a juvenile voice. Penile length was 4 cm, and testicular volume was 8 ml.
The LH level was not detectable; FSH was normal; testosterone, ACTH, and estradiol levels were low; and inhibin B, prolactin, IGF-I, cortisol, and TSH were normal. After iv administration of GnRH (100 ?g), the patient's level of FSH rose from 8.7 to 10.7 mIU/ml at 60 min, whereas LH remained undetectable. Serum testosterone increased after 5000 IU human chorionic gonadotropin administration from 0.49 to 4.32 ng/ml. The patient had azoospermia with low semen volume (0.2 ml). Magnetic resonance imaging of the brain and pituitary gland showed no abnormalities.
A diagnosis of hypogonadism due to selective LH deficiency was made. Treatment with im testosterone (250 mg every 3 wk) (Sustanon; Organon, Roseland, NJ) was initiated. Over a 6-month period, the testosterone induced virilization, penile growth to 8 cm in length, and an increase in testicular volume to 18 ml. During testosterone replacement therapy, the plasma levels of testosterone and 17?-estradiol increased, reaching the normal adult range (5.8 ± 1.0 ng/ml and 30 ± 1 pg/ml, respectively; mean ± SD of four determinations), LH remained undetectable. All the other hormones, assayed on three to four occasions, were within the normal range.
The treatment with testosterone was discontinued because of worsening of gynecomastia, and the therapy was shifted to chorionic gonadotropin 2000 IU three times a week (Gonasi HP; Amsa, Como, Italy). Although the treatment was irregular because of poor adherence, after 12 months the testicular volume was 18.6 ml, testosterone secretion was maintained within the normal adult range (4.92 ± 0.98 ng/ml; mean ± SD of six determinations), and the patient became oligospermic (0.38 × 106spermatozoa per ejaculate; semen volume, 0.38 ml), although the spermatozoa predominantly had abnormal shape and motility. All the other hormones studied were within the adult normal range except for LH, which remained undetectable.
In response to the administration of chorionic gonadotropin, the patient had a further increase in testicular volume and an enhanced production of testosterone, suggesting that the few Leydig cells observed in the testicular biopsy, although reduced in number and size, remained capable of steroidogenesis. Furthermore, the therapy with chorionic gonadotropin allowed for some spermatogenesis to take place. Thus, the absence of exposure to endogenous LH only partially altered the subsequent capacity for spermatogenesis. This treatment success improved the quality of life of the patient substantially.
This report expands the understanding of the genetics of hypogonadism and the knowledge of the mutation spectrum in isolated hypogonadotropic hypogonadism. Both affected siblings were compound heterozygous, whereas their unaffected parents were heterozygous and had a normal phenotype. This indicates that one copy of the LH ?-subunit gene is sufficient for normal LH secretion and function of the gonadotropic axis, ruling out haploinsufficiency. In conclusion, the LH ?-subunit mutations reported here are probably rare; however, they should be considered in girls with oligomenorrhea and boys with delayed puberty and selective deficiency of LH.
Basciani S, Watanabe M, Mariani S, et al. Hypogonadism in a Patient with Two Novel Mutations of the Luteinizing Hormone ?-Subunit Gene Expressed in a Compound Heterozygous Form. Journal of Clinical Endocrinology & Metabolism 2012;97(9):3031-8. Hypogonadism in a Patient with Two Novel Mutations of the Luteinizing Hormone ?-Subunit Gene Expressed in a Compound Heterozygous Form
Context: LH gene mutations are rare; only four mutations have been described. The affected individuals are hypogonadal.
Patient: We describe the clinical features of a 31-yr-old man who presented with delayed puberty and azoospermia and was found to have hypogonadism associated with an absence of circulating LH.
Main Outcome Measures and Results: The patient had a 12-bp deletion in exon 2 in the LH?-subunit gene and a mutation of the 5? splice site IVS2+1G?T in the same gene present in a compound heterozygous state. The first mutation predicts a deletion of four leucines of the hydrophobic core of the signal peptide. The second mutation disrupts the splicing of mRNA, generating a gross abnormality in the processing. The patient's heterozygous parents were clinically normal. The phenotype of a 16-yr-old sister of the proband, carrying the same mutations, was characterized by normal pubertal development and oligomenorrhea.
Conclusion: This report unravels two novel mutations of the LH gene critical for synthesis and activity of the LH molecule. The insight gained from the study is that normal pubertal maturation in women can occur in a state of LH deficiency, whereas LH is essential for maturation of Leydig cells and thus steroidogenesis, puberty, and spermatogenesis in man. These mutations should be considered in girls and boys with selective deficiency of LH.








 )]
)]